") 機(jī)器學(xué)習(xí)告訴你,新型冠狀病毒接下來(lái)將如何變異?
機(jī)器學(xué)習(xí)告訴你,新型冠狀病毒接下來(lái)將如何變異?
提起達(dá)爾文的生物進(jìn)化論,在人們的普遍認(rèn)知中,這是開(kāi)創(chuàng)現(xiàn)代科學(xué)的重要理論之一。像地球上其他所有為生存而掙扎的生物一樣,病毒也會(huì)進(jìn)化或變異。讓我們看看人類(lèi)病毒的來(lái)源——蝙蝠病毒的RNA核苷酸序列片段:AAAAT CAAA GCTT GTGTT GAA GAA GTTACAA CAACTCT GGAAG AAACTAAGTT與一小段人類(lèi)的新型冠狀病毒肺炎(Corona Virus Disease 2019,COVID-19)的RNA核苷酸序列:AAAAT TAAG GCTT GCATT GAT GAG GTTACCA CAACACT GGAAG AAACTAAGTT顯然,冠狀病毒已經(jīng)改變了它的內(nèi)部結(jié)構(gòu)以適應(yīng)新的宿主物種(更準(zhǔn)確地說(shuō),大約20%的冠狀病毒內(nèi)部結(jié)構(gòu)都發(fā)生了突變),但仍然保持了足夠數(shù)量的一致,使它仍然忠于它的起源物種。事實(shí)上,研究表明,COVID-19會(huì)不斷發(fā)生變異,以提高其存活率。在與冠狀病毒的對(duì)抗中,我們不僅需要探究擊敗病毒的方法,更需要明白病毒是如何變異的,以及如何應(yīng)對(duì)病毒變異。這篇文章中將從以下幾個(gè)方面進(jìn)行闡述:①?gòu)谋砻嫔辖忉孯NA核苷酸序列是什么②使用K-Means創(chuàng)建基因組信息集群③使用PCA實(shí)現(xiàn)可視化集群什么是基因組序列?DNA是脫氧核酸的簡(jiǎn)稱(chēng),其基本單位是脫氧核糖核苷酸(也叫脫氧核苷酸),是大多數(shù)生物的遺傳物質(zhì),在真核生物、原核生物、DNA病毒內(nèi)都存在的一種核酸;RNA則是核糖核酸的簡(jiǎn)稱(chēng),其基本單位是核糖核苷酸,是RNA病毒的遺傳物質(zhì)。新型冠狀病毒的基因序列就是RNA.基因組測(cè)序,通常被比作“解碼”,是分析取自樣本的脫氧核糖核酸(DNA)的過(guò)程。在每個(gè)正常細(xì)胞中有23對(duì)染色體,DNA的結(jié)構(gòu)是這樣的:
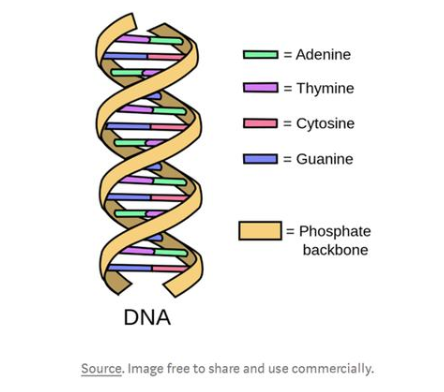
DNA卷曲的雙螺旋結(jié)構(gòu)可以使它展開(kāi)成階梯狀,這個(gè)梯子是由成對(duì)的化學(xué)字母組成的,叫做堿基。在DNA中有四種堿基:腺嘌呤、胸腺嘧啶、鳥(niǎo)嘌呤和胞嘧啶。腺嘌呤只與胸腺嘧啶結(jié)合,鳥(niǎo)嘌呤只與胞嘧啶結(jié)合,這些堿基分別用A、T、G和C表示。這些堿基形成了各種各樣的代碼,指導(dǎo)有機(jī)體如何構(gòu)建蛋白質(zhì)——這就是DNA如何控制病毒一舉一動(dòng)的基礎(chǔ)。

使用專(zhuān)門(mén)的設(shè)備,包括測(cè)序儀器和專(zhuān)門(mén)的標(biāo)簽,可以顯示特定的DNA序列片段。由此獲得的信息將經(jīng)過(guò)進(jìn)一步的分析和比較,使研究人員能夠識(shí)別基因的變化,與疾病和表型的關(guān)系,并確定潛在的藥物靶標(biāo)。一長(zhǎng)串的基因組序列A、T、G和C,代表了有機(jī)體對(duì)環(huán)境的反應(yīng),而生物體的突變又是通過(guò)改變DNA產(chǎn)生的,因此觀察基因組序列是分析冠狀病毒突變的有效手段,其中序列對(duì)齊法是常用的方法,主要通過(guò)將兩個(gè)或多個(gè)核酸序列或者蛋白質(zhì)序列進(jìn)行對(duì)比,并將其中相似的結(jié)構(gòu)區(qū)域突出顯示。序列對(duì)齊:給定兩個(gè)DNA序列A和B,對(duì)齊的方式是將空格分別插入到A和B序列中,得到具有相同長(zhǎng)度的對(duì)齊后的序列C和D;空格可以插入到任意的位置(包括兩端),但是相同位置不能同時(shí)為空格,也即是不存在C[i]和D[i]同時(shí)為空格的情況。然后為對(duì)齊后的序列的每個(gè)位置打分,總分為每個(gè)位置得分之和,具體的打分規(guī)則如下:a、如果C[i] == D[i]且都不是空格,得3分;b、如果C[i] != D[j]且都不是空格,得1分;c、如果C[i] 或者D[i]是空格,得0分。求給定原序列A和B的一個(gè)對(duì)齊方案,使得該對(duì)齊方案的總分最高。例如,序列原序列A和B如下:String strA = “GATC”; String strB = “ATCG”;則其中一個(gè)對(duì)齊方案如下:GATC**ATCG該方案總得分score=2*0+3*3 = 9分。因此,經(jīng)常通過(guò)序列對(duì)齊方式來(lái)比較序列與已知(尤其是功能和結(jié)構(gòu)已知的序列)之間的同源性,預(yù)測(cè)未知序列的功能。因此本文后續(xù)對(duì)于序列的分析主要是針對(duì)序列對(duì)齊后形成的指標(biāo)特征進(jìn)行探索和分析。數(shù)據(jù)的獲取數(shù)據(jù)可以在Kaggle上找到,如下圖所示:

每一行代表蝙蝠病毒的一個(gè)突變。首先,花一分鐘來(lái)欣賞大自然是多么不可思議——在幾周內(nèi),冠狀病毒已經(jīng)產(chǎn)生了262個(gè)突變來(lái)增加存活率。一些重要的列名解釋?zhuān)?/p>
query acc.ver表示原始的病毒標(biāo)識(shí)符。
subject acc.ver是病毒突變的標(biāo)識(shí)符。
% identity表示序列中與原始病毒相同的百分比。
Alignment length表示序列中有多少項(xiàng)是相同的或?qū)R的。
mismatches表示突變項(xiàng)和原始項(xiàng)之間的不同項(xiàng)數(shù)。
bit score代表了一個(gè)衡量標(biāo)準(zhǔn),衡量序列的對(duì)齊程度;分?jǐn)?shù)越高,對(duì)齊程度越高。
每一列的統(tǒng)計(jì)度量如下所示(這些可以在Python中運(yùn)用data.describe()語(yǔ)句被方便地調(diào)用):

有趣的是,通過(guò)查看% identity列,我們可以看到一個(gè)突變與原始病毒的最小對(duì)齊比率約為77.6%。然而巨大的標(biāo)準(zhǔn)偏差(7%的% identity)意味著原始病毒存在廣泛的變異范圍。在bit score中巨大標(biāo)準(zhǔn)偏差證實(shí)可以證實(shí)這一點(diǎn)——標(biāo)準(zhǔn)偏差大于平均值(即代表變異系統(tǒng)大于1,進(jìn)一步說(shuō)明了突變發(fā)生情況的多樣性)!通過(guò)相關(guān)性熱力圖可以很好的呈現(xiàn)變量之間的相關(guān)性,圖形中每個(gè)單元表示一個(gè)特征與另一個(gè)特征的相關(guān)性。

我們不難發(fā)現(xiàn),很多數(shù)據(jù)都是高度相關(guān)的,這是可以解釋的,因?yàn)榇蠖鄶?shù)的度量彼此存在一定的依賴性,因此導(dǎo)致變量之間存在高相關(guān)性,可以發(fā)現(xiàn)alignment length與bit score之間就具有高度相關(guān)性(0.94)。
使用K-Means來(lái)創(chuàng)建突變集群K-Means是一種聚類(lèi)算法,是通過(guò)機(jī)器學(xué)習(xí)的方式在特征空間中確定數(shù)據(jù)點(diǎn)相似群組。我們運(yùn)用K-Means的目標(biāo)是找到突變的群體,這樣我們就可以對(duì)突變的本質(zhì)以及如何針對(duì)性的處理它們有深入的了解。在此之前,我們首先需要確定集群k的數(shù)量,雖然這就像在二維空間中繪制一個(gè)點(diǎn)一樣簡(jiǎn)單,但在高維空間中是幾乎無(wú)法實(shí)現(xiàn)的(如果我們想要保留最多的信息)。若用“肘部法則”來(lái)選擇k會(huì)顯得過(guò)于主觀,且不準(zhǔn)確,所以我們會(huì)用輪廓法來(lái)代替。輪廓法是給不同取值k的集群打分,來(lái)區(qū)分聚類(lèi)的結(jié)果好壞程度(好的聚類(lèi):內(nèi)密外疏,同一個(gè)聚類(lèi)內(nèi)部的樣本要足夠密集,不同聚類(lèi)之間樣本要足夠疏遠(yuǎn))。Python中的sklearn庫(kù)將使K-Means和輪廓法的實(shí)現(xiàn)變得非常簡(jiǎn)單。
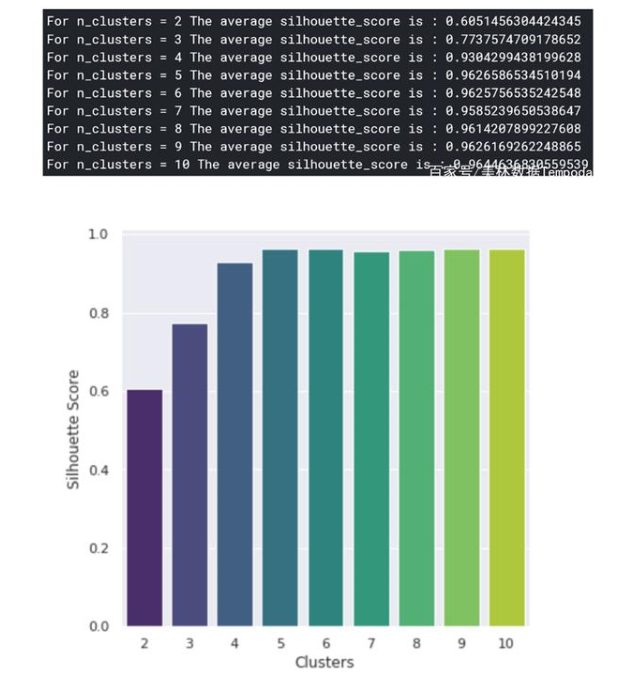
通過(guò)對(duì)上圖進(jìn)行分析,可以發(fā)現(xiàn)群體數(shù)為5時(shí)聚類(lèi)效果最佳。現(xiàn)在,我們可以進(jìn)一步確定群體中心,這些點(diǎn)是每個(gè)群體的中心,代表了不同群體的突變樣本的共性特征。

注:特征已經(jīng)被標(biāo)準(zhǔn)化,列與列之間無(wú)可比性
在此熱力圖中,行:代表不同的群體,列:代表每個(gè)群體的屬性。因?yàn)樵诰垲?lèi)之間需要對(duì)于特征按比例進(jìn)行縮放,以減少不同特征尺度差異的影響,所以圖中的數(shù)值在數(shù)量(縮放值,非原始尺度下的值)上沒(méi)有任何意義,但是,我們可以通過(guò)比較每個(gè)列中的縮放值,這使得我們可以對(duì)每個(gè)突變?nèi)后w的特征相對(duì)大小產(chǎn)生一個(gè)更直觀的感覺(jué)。通過(guò)對(duì)以上聚類(lèi)結(jié)果的分析,可以讓科學(xué)家將更多精力聚焦在對(duì)不同突變?nèi)后w的特征研究上,進(jìn)而針對(duì)性的研究不同類(lèi)型的疫苗,治療和預(yù)防也將變的更有目標(biāo)性。聚類(lèi)的結(jié)果已經(jīng)可以幫助我們解決很多方面的問(wèn)題,但由于存在高維特征及特征之間相關(guān)性的存在,讓我們不能更好的去解讀聚類(lèi)結(jié)果,因此,在下一節(jié)中,我們將使用PCA來(lái)實(shí)現(xiàn)聚類(lèi)結(jié)果的可視化呈現(xiàn)。利用PCA進(jìn)行集群可視化主成分分析是一種降維方法。它選擇多維空間中的正交向量來(lái)表示坐標(biāo)軸,通過(guò)特征的空間變換,可以有效降低特征之間的相關(guān)性,進(jìn)而通過(guò)貢獻(xiàn)度來(lái)保留最多的信息的特征,實(shí)現(xiàn)降維目的。同樣,我們可以通過(guò)Python的sklearn庫(kù),PCA的執(zhí)行可以被兩行代碼實(shí)現(xiàn)。首先,我們可以檢查被解釋的方差比(explained variance ratio),這是從原始數(shù)據(jù)集中保留的統(tǒng)計(jì)信息的百分比。在本例中,被解釋的方差比是0.9838548580740327,代表信息只有很少部分遺失!在此我們可以確信,無(wú)論我們從PCA得到什么分析,數(shù)據(jù)都是具有很高的可信度。每個(gè)新的特征(主成分)都是其他幾個(gè)列的線性組合。通過(guò)熱力圖,我們可以直觀地了解每一特征對(duì)于兩個(gè)成份(新的特征)中的重要性。

通過(guò)以上圖中數(shù)值的分析,關(guān)鍵是要理解在成分1中出現(xiàn)高數(shù)值是什么意思——在這種情況下,它的特點(diǎn)是有著更高的一致性,即更接近原始病毒;成分2的主要的特點(diǎn)是擁有更低的一致性,即突變遠(yuǎn)離原始值,這也反映在bit score的較大差異上。

通過(guò)主成分將所有樣本映射到2維空間體系下,可以很明顯發(fā)現(xiàn),病毒突變有5條主線,以下通過(guò)對(duì)這5條線的分析,可以讓我們獲取更多的信息。可以發(fā)現(xiàn),有四個(gè)病毒突變?cè)诘谝恢鞒煞郑╔軸)的左邊,一個(gè)在右邊。第一主成分的特征是alignment length具有很高的取值,這意味著第一個(gè)主成分的值越高,對(duì)應(yīng)的alignment length就越長(zhǎng)(越接近原始病毒)。因此,第一主成分的低值區(qū)與原始病毒的遺傳距離較遠(yuǎn),即大多數(shù)病毒集群與原始病毒有很大不同。因此,試圖研制疫苗的科學(xué)家應(yīng)該意識(shí)到,這種病毒會(huì)發(fā)生大量變異。第二主成分(Y軸)在同一群體之間的差異性很小,在不同群體之間明顯分為3個(gè)區(qū)段,這就需要后續(xù)我們進(jìn)一步分析,以便能夠更好的對(duì)于突變?nèi)后w進(jìn)行深入了解。結(jié)論本文一方面通過(guò)使用K-Means聚類(lèi)算法,能夠幫助我們從眾多突變樣本中快速識(shí)別冠狀病毒的五個(gè)主要典型突變?nèi)后w,另一方面用PCA分析方法在二維空間中實(shí)現(xiàn)這些群體的可視化展現(xiàn),通過(guò)展示結(jié)果可以很直觀的呈現(xiàn)冠狀病毒有很高的突變率(這可能就是它如此致命的原因),通過(guò)對(duì)于這些分析結(jié)果,對(duì)于研制冠狀病毒疫苗的科學(xué)家來(lái)說(shuō),可以利用群體的共性特征值結(jié)合領(lǐng)域?qū)I(yè)知識(shí)來(lái)充分解讀每個(gè)群體的特征信息,以便有針性的、更好的指導(dǎo)疫苗的研制及預(yù)防工作。
-
DNA
+關(guān)注
關(guān)注
0文章
243瀏覽量
31058 -
K-means
+關(guān)注
關(guān)注
0文章
28瀏覽量
11316
發(fā)布評(píng)論請(qǐng)先 登錄
相關(guān)推薦
芯系武漢半導(dǎo)體企業(yè)向受冠狀病毒地區(qū)捐贈(zèng)匯總
怎樣預(yù)防手機(jī)上的新型冠狀病毒傳播
新型冠狀病毒加速了“智慧城市”對(duì)“傳統(tǒng)城市”的代替
預(yù)防新型冠狀病毒需要什么智能設(shè)備應(yīng)該如何使用
新型冠狀病毒對(duì)Uber業(yè)務(wù)影響大
蘋(píng)果和微軟這些美國(guó)科技巨頭是怎么應(yīng)對(duì)新型冠狀病毒的
Google Maps如何幫助解決冠狀病毒的流行
美國(guó)擬啟用Summit超算系統(tǒng)制止新型冠狀病毒大流行的蔓延
波音公司已持有150億美元現(xiàn)金來(lái)渡過(guò)新型冠狀病毒疫的危機(jī)
利用機(jī)器學(xué)習(xí)發(fā)現(xiàn)新型冠狀病毒的潛在中和抗體
聊天機(jī)器人將如何解決冠狀病毒帶來(lái)的人員配備問(wèn)題
淺談3D打印技術(shù)在抗擊新型冠狀病毒疫情中的應(yīng)用
同茂線性馬達(dá)談?dòng)蛛p變異的新型冠狀病毒
社交距離提醒(冠狀病毒)開(kāi)源案例

冠狀病毒實(shí)時(shí)更新器開(kāi)源分享





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論