 DPO-TXO2如何讀取數據用于結果分析
DPO-TXO2如何讀取數據用于結果分析
鴻之微BDF(Beijing Density Functional)軟件是一個獨立完整、具有完全自主知識產權的量子化學計算軟件包,也是國際上第一個基于現代密度泛函理論、能準確計算分子體系基態總能量的完全相對論密度泛函程序。
BDF的研發始于1993年,并于1997年正式命名。BDF對稀土、錒系、超重元素的計算結果一直被作為檢驗其他近似相對論方法的基準。BDF對重元素體系電子、分子結構的計算結果被后續20余個實驗驗證。
熱激活延遲熒光(TADF)材料是繼熒光材料和貴金屬磷光材料之后發展起來的第三代純有機延遲熒光材料,其典型的特征是較小的單三重態能隙(ΔES-T)和溫度正相關性。
2012年,日本九州大學的Chiahaya Adachi課題組首次報道外量子效率(EQE)超過20%的4CzIPN 分子[ i ],該材料的單線態和三線態能級差幾乎為0,在室溫下(298 K)的這樣的熱擾動下激子完全能夠從三線態再回到單線態而發射熒光,因此命名為TADF(Thermally activated delayed fluorescence)。
當S1與T1的激發都是HOMO-》LUMO特征,二者的能量差為2*K,K是HOMO與LUMO間的交換積分。隨著HOMO與LUMO分離的增加,K會迅速減小。所以分離較大的時候,S1與T1 gap就較小,易于發生TADF需要的RISC。
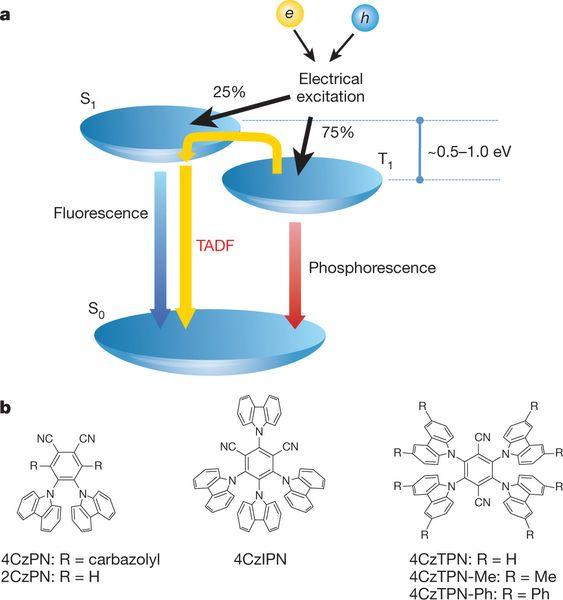
為了保證高效的RISC,TADF材料需要具有較小的單三重態能隙,對應其HOMO/LUMO的有效分離,因此,TADF材料一般采用給體(D)?受體(A)、D?A?D的結構以不同的給受體作用實現HOMO/LUMO分離,同時兼顧其躍遷振子強度。
不同給受體的電子特性、三重態能級、結構剛性及扭曲程度等均均會影響材料的△EST、振子強度、態密度、激子壽命等,最終反映在材料的光物理性能和對應OLED器件的光電性能上。
本專題將以一個典型的TADF分子DPO-TXO2為例,介紹如何計算結構優化、頻率、單點能、激發能、自旋軌道耦合等。同時介紹如何讀取數據用于結果分析,幫助用戶深入了解BDF軟件的使用。
一、結構優化和頻率計算
1、生成結構優化和頻率輸入文件
在Device Studio中導入準備的分子結構DPO-TXO2.xyz得到如圖1.1-1所示界面,選中 Simulator → BDF → BDF,在彈出的界面中設置參數。計算結構優化時計算類型選擇Opt+Freq,方法、泛函、基組等選項用戶可根據計算需要設置參數。例如Basic Settings面板設置為圖1.1-2,SCF面板消除“Use MPEC+COSX”勾選(圖1.1-3)、OPT 、Freq面板仍為默認值,之后點擊 Generate files 即可生成對應計算的輸入文件。生成的輸入文件 bdf.inp參數部分如圖1.1-4所示 ,此時Device Studio圖形界面如圖1.1-5所示。
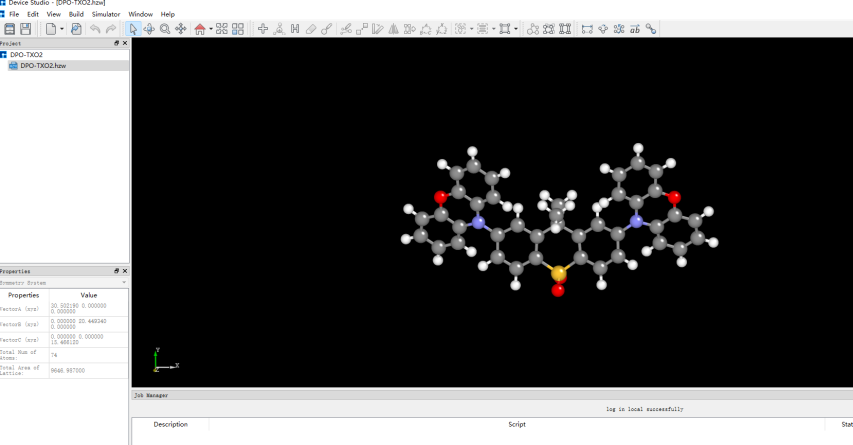
圖1.1-1
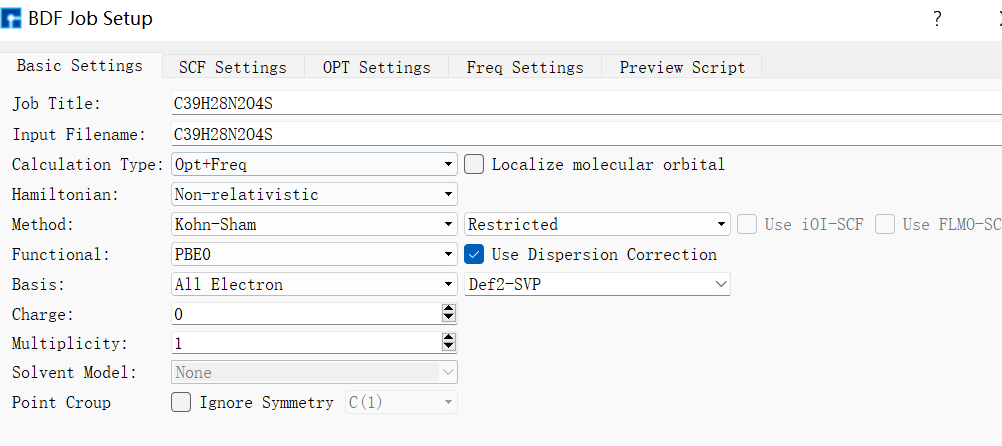
圖1.1-2
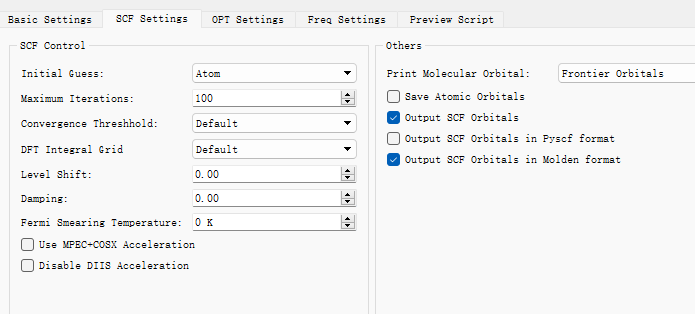
圖1.1-3

圖1.1-4
圖1.1-5
備注:此處為保證結構優化和頻率計算的條件相同,計算類型選擇Opt+Freq,可以的單獨做Opt計算或Freq計算。
2、BDF計算
在做BDF計算之前,需連接裝有BDF的服務器,具體配置過程見鴻之微云操作指南。連接好服務器,在做計算之前,用戶可根據需要打開輸入文件并查看文件中的參數設置是否合理,若不合理,則可選擇直接在文件中編輯或重新生成,再進行BDF計算。
在圖1.1-5所示的界面中,選中 bdf.inp → 右擊 → Run,在彈出的界面導入相應的腳本,點擊Run提交作業,如圖1.1-6。計算完成后點擊下載按鈕彈出計算結果界面如圖1.1-7所示,選擇.out結果文件,點擊 Download下載。(提交作業操作為重復內容,在后面的計算中將不再贅述)
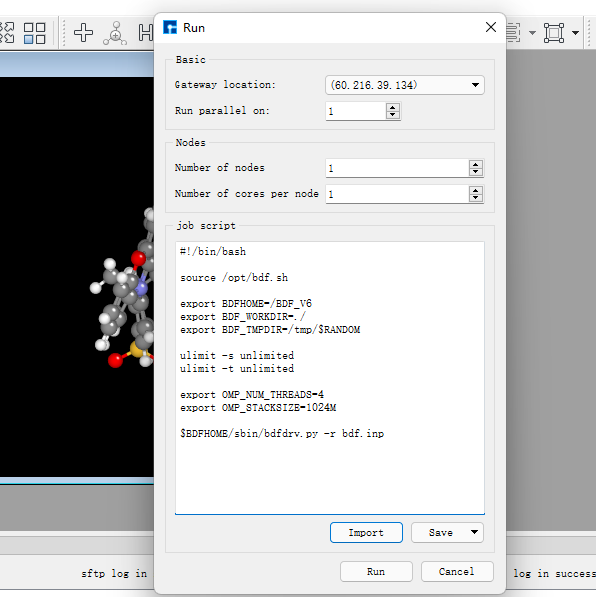
圖1.1-6
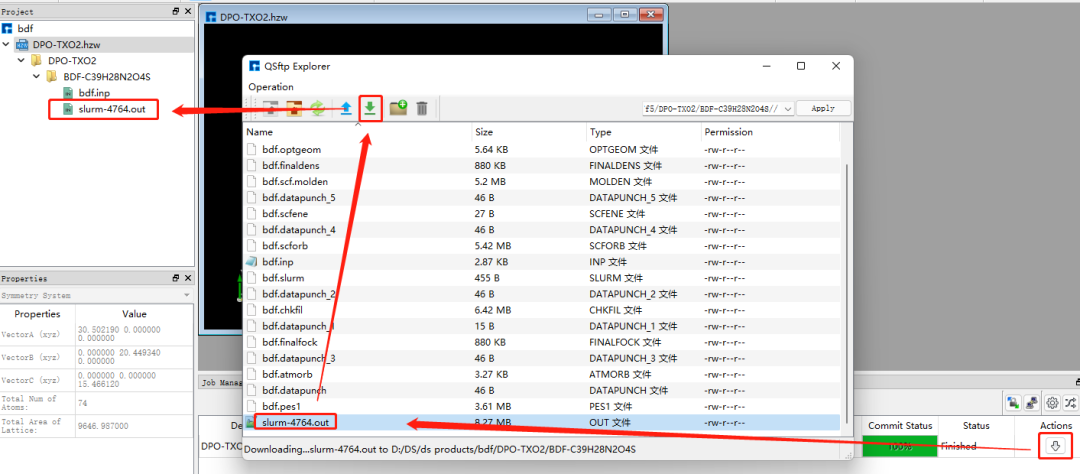
圖1.1-7
3、結構優化結果分析
右擊下載后的out文件,選擇Open with/Open containing folder即可查看結果文件。找到如圖1.1-8所示部分,當Geom.converge的4個值均為YES時,證明結構優化收斂。上方和下方分別為收斂的分子結構笛卡爾坐標和內坐標。優化后的坐標信息可以作為初始結構用于后續計算。

圖1.1-8
檢查頻率,若不存在虛頻證明結構穩定。
二、單點能計算

1、生成單點能輸入文件
將優化后的坐標導入Device Studio,名字改為DPO-TXO2-sp.xyz,此時圖形界面如圖1.2-1。
圖1.2-1
選中 Simulator → BDF → BDF,在彈出的界面中計算類型選擇Single Point(默認值),方法、泛函、基組等選項用戶可根據計算需要設置參數。例如泛函選PBE0,基組Def2-TZVP,其他參數仍為默認值,之后點擊 Generate files 即可生成對應計算的輸入文件。生成的輸入文件 bdf.inp參數部分如圖1.2-2所示。
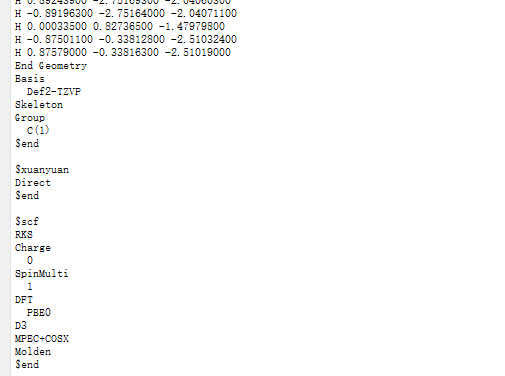
圖1.2-2
2、BDF計算
同結構優化計算相同,連接好裝有BDF的服務器后,選中 bdf.inp → 右擊 → Run,檢查腳本沒有問題,點擊Run提交作業。計算完成后點擊下載按鈕彈出計算結果,選擇.out結果文件,點擊 Download下載。
3、單點能結果分析
右擊下載后的out文件,選擇Open with/Open containing folder即可查看結果文件。找到E_tot為系統總能量(圖1.2-3),E_tot=E_ele + E_nn,本例中系統總能量為-2310.04883102 Hartree。E_ele是電子能量,E_nn是原子核排斥能,E_1e是單電子能量,E_ne 是原子核對電子的吸引能,E_kin 是電子動能,E_ee 是雙電子能,E_xc 是交換相關能。
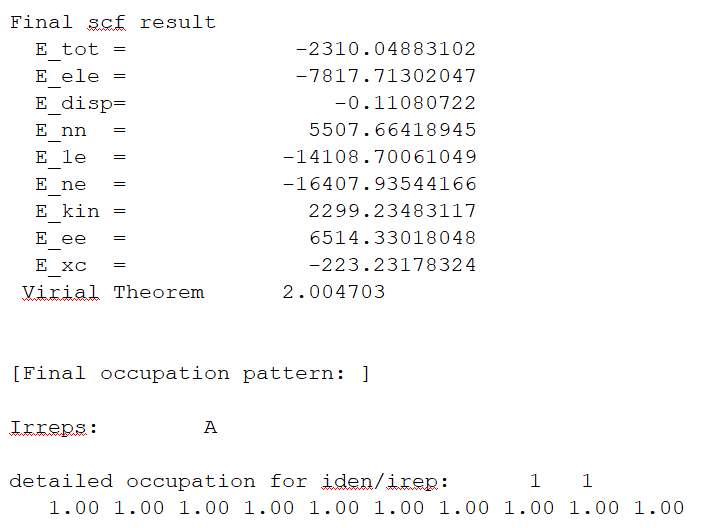
圖1.2-3
下方為軌道的占據情況,以及軌道能、HOMO-LUMO gap等信息,如圖1.2-4。HOMO為-5.358 eV,LUMO為-1.962 eV,HOMO-LUMO gap為3.396 eV,Irrep為不可約表示,代表分子軌道對稱性,本例中HOMO、LUMO不可約表示序號均為A。

圖1.2-4
最底部為Mulliken和Lowdin電荷布局、偶極矩信息。圖1.2-5為部分截取。
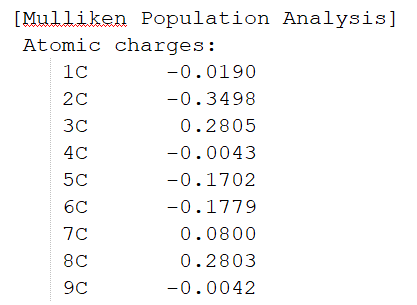
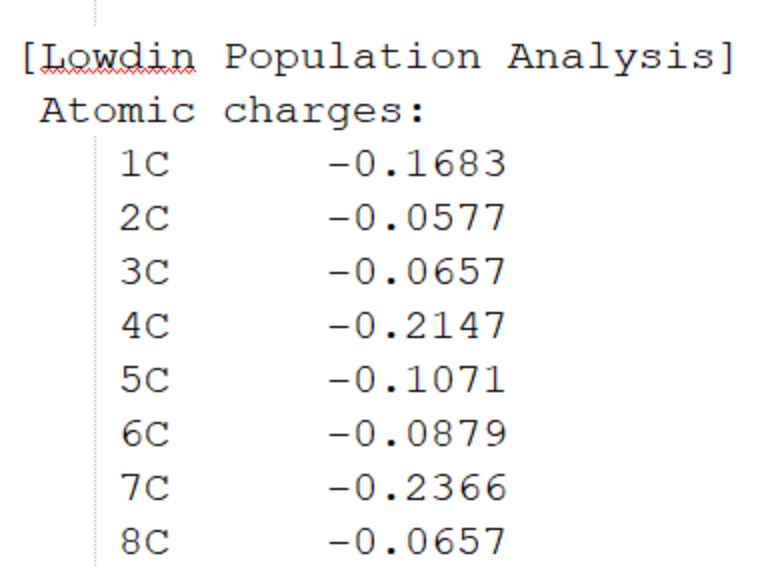

圖1.2-5
4、查看HOMO軌道圖
為了更清楚的了解電子結構,往往需要做前線分子軌道分析。目前發布的版本BDF2022A中還無法實現數據的后處理,HOMO、LUMO軌道圖可以用第三方軟件Multiwfn+VMD渲染,需要用到scf.molden文件,軟件的使用方法在量化論壇有專門的帖子可以學習,此文不作涉及。
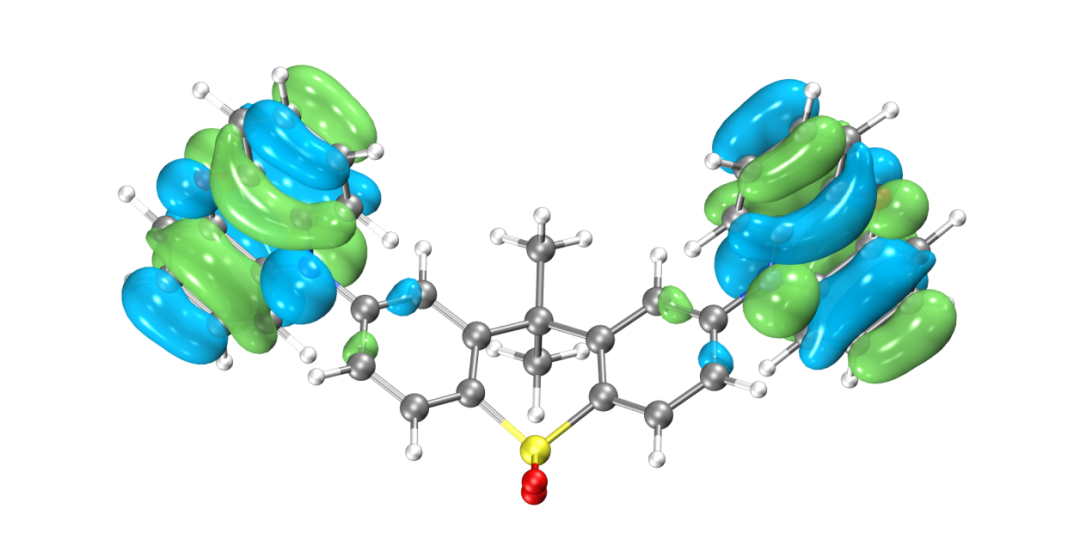
HOMO軌道分布圖
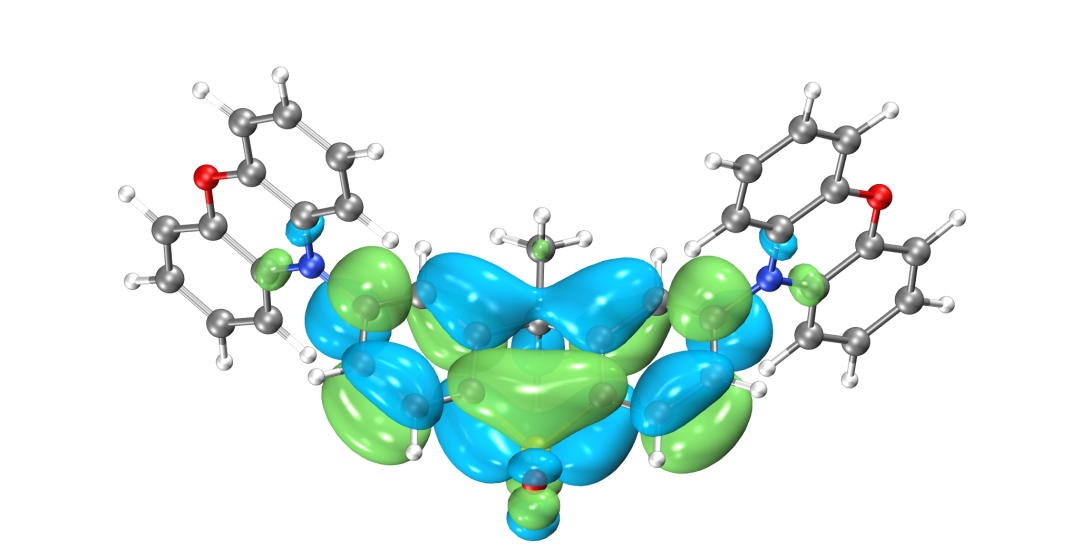
LUMO軌道分布圖
得到的最高占據軌道(HOMO)與最低非占據軌道(LUMO)如圖所示,由于兩側對稱分布的吩惡嗪雜環是一個典型的給電子結構,而中心的磺酰化的四氫化萘是一個典型的吸電子的結構,因此整個分子是非常典型的D-A-D結構。可以看到HOMO軌道主要分布在兩翼,LUMO軌道分布在中心,HOMO和LUMO軌道幾乎沒有重疊,符合TADF分子的電子結構特征。當然并不是所有HOMO和LUMO軌道分離的分子都具有TADF的光電特性,還需要滿足S1和T1激發都是HOMO-》LUMO軌道躍遷才行,因此我們可以進一步用BDF軟件計算該分子的激發態電子結構。
-
數據
+關注
關注
8文章
7033瀏覽量
89040 -
頻率
+關注
關注
4文章
1500瀏覽量
59229 -
OLED器件
+關注
關注
0文章
9瀏覽量
10160
原文標題:鴻之微BDF軟件計算賞析|理論揭示DPO-TXO2的熱激活延遲熒光(TADF)發光機制(一)
文章出處:【微信號:hzwtech,微信公眾號:鴻之微】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦






 工商網監
工商網監
評論