") 體相擴(kuò)散率和界面形貌對(duì)金屬負(fù)極剝離容量的影響
體相擴(kuò)散率和界面形貌對(duì)金屬負(fù)極剝離容量的影響
研究背景
作為鋰離子電池負(fù)極材料,金屬鋰因其高比容量(3860 mAh·g-1)而備受關(guān)注。然而,鋰的體相擴(kuò)散率往往是決定循環(huán)行為的一個(gè)限制因素。特別是,在固態(tài)電池循環(huán)過程中,需要鋰原子的快速體擴(kuò)散來維持金屬鋰電極與固體電解質(zhì)之間的界面接觸。
鋰合金的使用有助于與固體電解質(zhì)形成更穩(wěn)定的界面。這通常歸因于鋰合金中鋰的快速擴(kuò)散。據(jù)報(bào)道,許多合金的鋰擴(kuò)散率顯著超過鋰自擴(kuò)散率。Mg在Li中的溶解度范圍非常寬(圖1a),因此,鋰鎂電極在電化學(xué)循環(huán)過程中可能不發(fā)生相變,從而有望提供更好的微觀結(jié)構(gòu)穩(wěn)定性。然而,Li-Mg合金中的鋰擴(kuò)散率值在文獻(xiàn)中存在差異。
成果簡(jiǎn)介
近日,牛津大學(xué)Chris R. M. Grovenor教授在ACS Energy Letters上發(fā)表了題為“On the Relative Importance of Li Bulk Diffusivity and Interface Morphology in Determining the Stripped Capacity of Metallic Anodes in Solid-State Batteries”的論文。該工作用同位素示蹤法研究了鋰在Li-Mg合金中的擴(kuò)散率,發(fā)現(xiàn)鎂的存在減緩了鋰的擴(kuò)散。
在大的剝離電流下,脫鋰過程是擴(kuò)散限制的,因此鋰金屬電極比鋰鎂電極產(chǎn)生更大的容量。然而,在較低的電流下,從鋰鎂電極中可以提取更多的鋰,證明合金可以保持一個(gè)更穩(wěn)定的擴(kuò)散路徑到固體電解質(zhì)表面,從而提高了鋰的有效擴(kuò)散率。
研究亮點(diǎn)
(1)本工作通過使用同位素示蹤法直接測(cè)量鋰在鋰鎂合金中的鋰擴(kuò)散率。為此,通過耦合兩種具有不同同位素濃度的材料來制備同位素異質(zhì)結(jié)構(gòu)。為了確保兩種材料之間的緊密接觸,使用熱蒸發(fā)將示蹤同位素薄膜沉積在不同同位素濃度的樣品上。圖1b顯示,安裝在等離子體聚焦離子束(PFIB)儀器中的二次離子質(zhì)譜(SIMS)探測(cè)器用于跟蹤同位素濃度隨深度的變化,從中可以得出擴(kuò)散系數(shù)。
(2)SIMS分析顯示,鋰在Li-Mg合金中的擴(kuò)散率比鋰在金屬鋰中的自擴(kuò)散率低約1個(gè)數(shù)量級(jí)。因此,使用鋰鎂合金負(fù)極使得固態(tài)電池性能改善不是因?yàn)殇嚁U(kuò)散率增加導(dǎo)致的。當(dāng)采用石榴石固體電解質(zhì)時(shí),金屬鋰在大剝離電流密度下優(yōu)于合金負(fù)極。然而,低溫PFIB切片研究顯示,合金負(fù)極與固體電解質(zhì)的接觸更穩(wěn)定,因此在較小的剝離電流密度和無外部壓力下能夠獲得更大的容量。
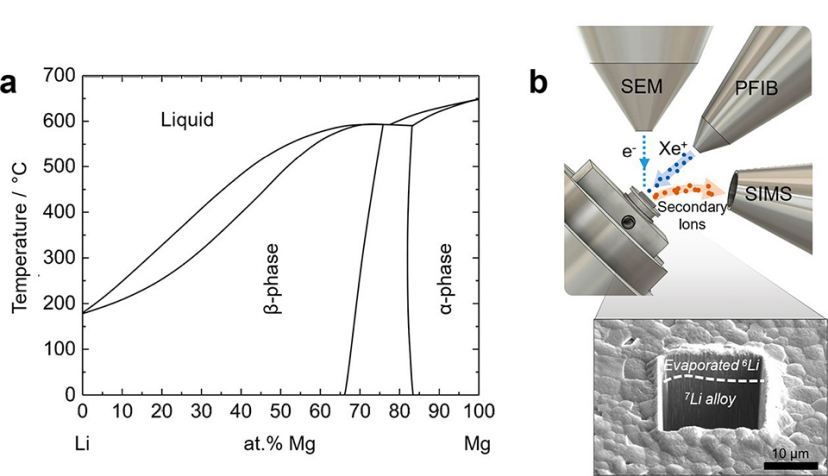
圖 1、(a)Li-Mg體系的平衡相圖。(b)用SIMS測(cè)量擴(kuò)散率的實(shí)驗(yàn)裝置。
圖文導(dǎo)讀
為了研究金屬鋰的自擴(kuò)散,將厚度為~5 μm的7Li薄膜熱蒸發(fā)到6Li上。為了測(cè)量7Li-Mg合金中的擴(kuò)散,6Li被蒸發(fā)到合金表面。圖2顯示了從熱蒸發(fā)開始60分鐘,同位素示蹤劑在不同基質(zhì)中的典型擴(kuò)散分布圖。
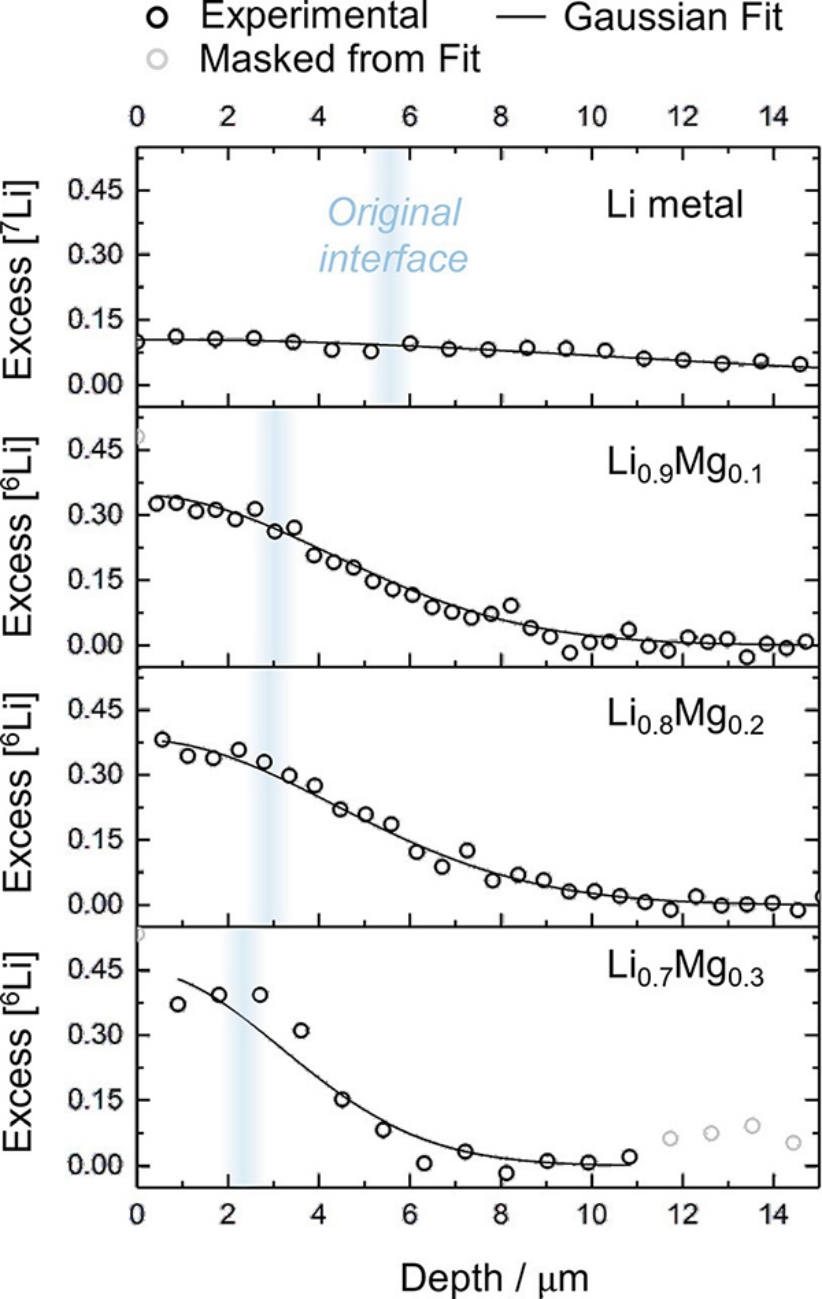
圖 2、從示蹤物沉積開始60分鐘后,示蹤同位素進(jìn)入基質(zhì)的過量濃度SIMS擴(kuò)散曲線。從頂部開始:7Li變成6Li,然后6Li變成7Li1–xMgx,x=0.1、0.2和0.3。通過橫斷面二次電子成像推斷出示蹤劑與襯底之間原始界面的位置。
根據(jù)圖2中7Li向6Li的擴(kuò)散分布,計(jì)算出鋰自擴(kuò)散(示蹤劑擴(kuò)散)系數(shù)DLi*為(1.6±0.1)×10-10cm2·s-1,與以往文獻(xiàn)報(bào)道基本一致。然而,鋰在鋰鎂合金中的擴(kuò)散率并沒有得到改善。基于圖2中的擴(kuò)散曲線,在10、20和30 at.%Mg時(shí),鋰的本征擴(kuò)散系數(shù)DLi分別為(2.4±0.1)、(2.6±0.1)和(1.4±0.2)×10–11cm2·s–1。這種較慢的擴(kuò)散率也可以在圖2中看到,在所有情況下,Li-Mg合金上示蹤同位素的表面濃度都大于金屬鋰。
Li-Mg合金中的β相是一種置換固溶體,由于Li和Mg的戈德施密特半徑只有幾個(gè)百分點(diǎn)的差異,因此可以假設(shè)鋰在Li-Mg中的擴(kuò)散機(jī)制與鋰單質(zhì)中相同,都是通過空位擴(kuò)散。金屬鋰和鋰鎂合金材料之間的另一個(gè)可能影響測(cè)量擴(kuò)散率的差異是晶粒尺寸,但所有材料的晶粒尺寸都在100 μm量級(jí),因此晶界對(duì)整體擴(kuò)散通量的貢獻(xiàn)應(yīng)該是相似的,可以忽略不計(jì)。
當(dāng)考慮鋰通量擴(kuò)散到Li-Mg合金中時(shí),存在一個(gè)成分梯度,該梯度會(huì)引入相關(guān)的活性梯度,但一般來說,向較低熔點(diǎn)基體A中添加較高熔點(diǎn)的元素B應(yīng)會(huì)降低DA的值。
測(cè)出的DLi值與文獻(xiàn)中基本一致,例如Korblein等人(使用核磁共振<7×10–11cm2·s–1)和Zhang等人(使用中子斷層掃描技術(shù)6×10–11cm2·s–1),以及Krauskopf等人(3×10–11cm2·s–1)對(duì)Li-Mg合金與固體電解質(zhì)接觸的研究。靜電位電化學(xué)滴定法測(cè)得的Li-Mg合金中鋰擴(kuò)散率更高,與文獻(xiàn)的差異較大,這可能是由于電化學(xué)方法得到的擴(kuò)散系數(shù)受合金微觀結(jié)構(gòu)及其表面形貌的影響,多孔結(jié)構(gòu)越大擴(kuò)散系數(shù)越大。
事實(shí)上,對(duì)于這種方法,計(jì)算擴(kuò)散系數(shù)需要知道合金的真實(shí)表面積。這些不確定性的來源會(huì)導(dǎo)致擴(kuò)散率誤差高達(dá)4個(gè)數(shù)量級(jí)。而這里使用的示蹤劑擴(kuò)散法能夠直接探測(cè)體擴(kuò)散率,而不需要模型假設(shè)。
根據(jù)Schmalzried和Janek的模型預(yù)測(cè),本研究中發(fā)現(xiàn)鋰的自擴(kuò)散率DLi*過低,即使在較小的電流密度(50-200 μA·cm-2)下,鋰電極也無法與固體電解質(zhì)保持形貌穩(wěn)定的界面,因此除非施加較大的外部壓力,否則在剝離過程中會(huì)形成空洞。鋰鎂合金中DLi值越小,說明鋰鎂合金負(fù)極的鋰化和剝離動(dòng)力學(xué)越慢。
通過將鋰和Li0.9Mg0.1電極與鋰石榴石固體電解質(zhì)Ta:LLZO接觸,并進(jìn)行剝離實(shí)驗(yàn)證實(shí)了這一點(diǎn)。在剝離過程中不施加外部壓力,即負(fù)極中的鋰通量只取決于鋰擴(kuò)散率,可以排除蠕變變形的影響。圖3a顯示了在1 mA·cm-2下的剝離電位曲線。當(dāng)固體電解質(zhì)表面的鋰濃度降低時(shí),電池的極化隨時(shí)間增加而增加。
用低溫PFIB對(duì)這些界面進(jìn)行橫切面分析(圖3b),結(jié)果表明,對(duì)于金屬鋰電極來說,界面形成了大的空隙,幾乎完全與電解質(zhì)失去接觸。然而,Li0.9Mg0.1合金電極能夠與固體電解質(zhì)保持更穩(wěn)定的接觸形態(tài)。鋰以相同的速度從兩個(gè)電極中剝離出來,盡管使用Li0.9Mg0.1很大程度上防止了大空隙的形成,但其較慢的體相鋰擴(kuò)散率(DLi)使得只能剝離有限容量的鋰。
有趣的是,如果Li0.9Mg0.1電極在第一次脫鋰后停留幾個(gè)小時(shí),由于維持了擴(kuò)散路徑,鋰可以擴(kuò)散回固體電解質(zhì)表面,在隨后的剝離步驟中可以獲得一些額外的容量(圖3a)。相比之下,鋰金屬電極和固體電解質(zhì)之間完全失去接觸,在沒有外部壓力的情況下無法恢復(fù)接觸,即使在休息期之后也無法繼續(xù)剝離。
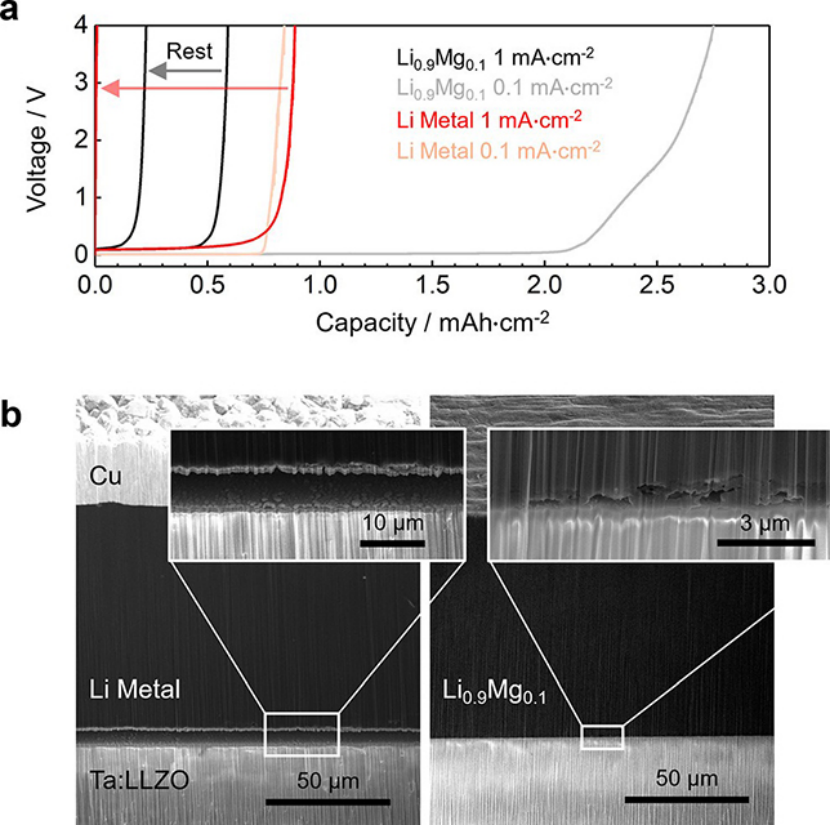
圖 3、(a)在1 mA·cm-2和0.1 mA·cm-2下,鋰和Li0.9Mg0.1電極的剝離實(shí)驗(yàn)。(b)在1 mA·cm-2下,工作電極剝離后的低溫PFIB截面二次電子圖像。
圖3a顯示,當(dāng)使用較小的剝離電流(0.1 mA·cm-2)時(shí),鋰金屬電極獲得了相似的容量,這表明在不施加外部壓力的情況下,在相同的剝離容量后形成了類似數(shù)量的空洞。然而,由于Li0.9Mg0.1能夠與固體電解質(zhì)保持良好的接觸,從Li0.9Mg0.1電極中脫出的容量是1 mA·cm-2下的4倍多。這一結(jié)果表明,如果脫鋰速率足夠低,鋰原子的體相擴(kuò)散可以向界面提供足夠的通量。
在實(shí)際運(yùn)行條件下,合金電極的鋰活性會(huì)發(fā)生變化,且很難預(yù)測(cè),這可能會(huì)改變鋰擴(kuò)散率(DLi應(yīng)取決于鋰的局部濃度)和電荷轉(zhuǎn)移效率。然而,從上面報(bào)道的結(jié)果和以前的研究中可以發(fā)現(xiàn),擴(kuò)散系數(shù)會(huì)隨著鎂含量的增加而減小,這與熔化溫度的急劇升高是一致的(見圖1a)。
然而,鋰鎂合金似乎能夠通過部分脫鋰的鋰鎂相,使負(fù)極主體中的鋰原子連續(xù)進(jìn)入固體電解質(zhì)表面,而鋰不會(huì)通過鋰電極中的空隙擴(kuò)散。因此,可以通過考慮有效鋰擴(kuò)散率來理解鋰鎂合金的行為,該擴(kuò)散率考慮了鋰的體擴(kuò)散率,以及不同的孔隙形態(tài)控制電極和固體電解質(zhì)之間接觸面積的方式。當(dāng)鋰金屬電極中形成空洞時(shí),其有效擴(kuò)散率迅速下降。鋰鎂電極具有較小的初始有效擴(kuò)散率,但在循環(huán)過程中界面形貌更穩(wěn)定,因此在小剝離速率下提高了容量。
在鋰擴(kuò)散率實(shí)驗(yàn)期間,應(yīng)注意盡量減少蒸發(fā)示蹤薄膜與6Li或7Li-Mg襯底之間的界面污染。然而,一些污染是不可避免的。為了了解其對(duì)鋰擴(kuò)散率的影響,圖4a比較了7Li到6Li的兩個(gè)擴(kuò)散曲線,其中6Li襯底是在7Li示蹤物沉積之前在具有不同污染程度的手套箱中制備的。
在含有~1ppm H2O和O2的手套箱中,用SIMS采集的18O信號(hào)顯示,原始7Li–6Li界面處有一個(gè)明顯的峰,表明控制較差的環(huán)境中,6Li表面受到污染。在這種情況下,測(cè)量到的擴(kuò)散系數(shù)比之前報(bào)道的值低一個(gè)數(shù)量級(jí),所以即使是少量的污染也會(huì)對(duì)測(cè)量到的鋰擴(kuò)散率產(chǎn)生很大的影響。
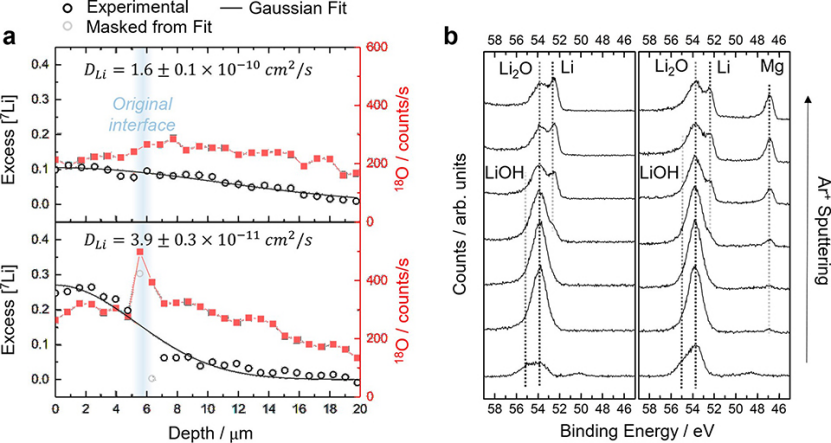
圖 4、(a)示蹤劑沉積開始后60分鐘,7Li向6Li的擴(kuò)散曲線,包括18O SIMS信號(hào)。上圖:在H2O和O2含量<0.1 ppm的手套箱中制備的6Li基底。下圖:在H2O和O2含量~1 ppm的手套箱中制備的6Li基底。(b)6Li(左)和7Li0.9Mg0.1(右)新鮮表面的Li 1s和Mg 2p XPS信號(hào)。采用Ar+進(jìn)行深度濺射。
另外,還通過XPS分析研究了6Li和Li-Mg表面的污染程度(圖4b)。即使是新鮮的表面也含有一些來自鋰氧化物和氫氧根的信號(hào),但在XPS室中進(jìn)行原位Ar+濺射可以揭示底層的金屬信號(hào)。在所有電極表面都可以看到相同的污染物種類。因此,在金屬鋰和鋰鎂合金上,表面污染對(duì)鋰擴(kuò)散率測(cè)量的影響應(yīng)該是相似的。
總結(jié)與展望
本工作利用同位素示蹤法研究了鋰在10、20和30 at.%的鋰鎂合金中的擴(kuò)散率,以及鋰的自擴(kuò)散率,其中采用SIMS跟蹤同位素?cái)U(kuò)散。結(jié)果表明,鋰在Li-Mg中的擴(kuò)散率(~2×10–11cm2·s–1)大大低于鋰的自擴(kuò)散率(1.6×10–10cm2·s–1)。盡管鋰鎂合金中鋰體擴(kuò)散率較小,但如果使用相對(duì)較小的電流密度,鋰鎂合金可以提供比鋰金屬電極更大的剝離容量。
這是因?yàn)長(zhǎng)i-Mg合金與固體電解質(zhì)能夠保持穩(wěn)定的界面接觸,從而使得鋰原子可以持續(xù)擴(kuò)散到電解質(zhì)表面。也就是說,界面形態(tài)的改善提高了運(yùn)行穩(wěn)定性。而鋰金屬電極中的大空隙阻礙了鋰原子的傳輸,即使負(fù)極中的體擴(kuò)散系數(shù)較大。然而,當(dāng)使用較大的電流密度時(shí),鋰鎂合金電極中較慢的體擴(kuò)散動(dòng)力學(xué)最終會(huì)限制剝離容量,因此在這些條件下,鋰金屬電極性能更好。
如果要在大電流密度下使用固態(tài)電池,那么尋找具有快速鋰體擴(kuò)散率的鋰合金仍然很重要。
審核編輯:劉清
-
鋰離子電池
+關(guān)注
關(guān)注
85文章
3243瀏覽量
77784 -
固態(tài)電池
+關(guān)注
關(guān)注
10文章
701瀏覽量
27850 -
固體電解質(zhì)
+關(guān)注
關(guān)注
0文章
46瀏覽量
8406
原文標(biāo)題:牛津大學(xué)ACS Energy Lett.:體相擴(kuò)散率和界面形貌對(duì)金屬負(fù)極剝離容量的影響
文章出處:【微信號(hào):清新電源,微信公眾號(hào):清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請(qǐng)注明出處。
發(fā)布評(píng)論請(qǐng)先 登錄
相關(guān)推薦
利用液態(tài)金屬鎵剝離制備二維納米片(2D NSs)的方法

篩選理想的預(yù)鋰化正極應(yīng)用于無負(fù)極金屬鋰電池

多功能高熵合金納米層實(shí)現(xiàn)長(zhǎng)壽命無負(fù)極鈉金屬電池

離子液體添加劑用于高壓無負(fù)極鋰金屬電池

導(dǎo)體和絕緣體的電阻率比較 電阻率檢測(cè)技術(shù)的發(fā)展趨勢(shì)
如何提高金屬探測(cè)器探測(cè)率
通過電荷分離型共價(jià)有機(jī)框架實(shí)現(xiàn)對(duì)鋰金屬電池固態(tài)電解質(zhì)界面的精準(zhǔn)調(diào)控
什么是滲透作用_金屬封裝又是如何發(fā)生滲透
全固態(tài)鋰金屬電池的鋰陽(yáng)極夾層設(shè)計(jì)
石墨負(fù)極在鋰離子電池中的發(fā)展與儲(chǔ)鋰機(jī)制
一種新型的鈉金屬電池負(fù)極穩(wěn)定化策略
外延片和擴(kuò)散片的區(qū)別是什么
具有分級(jí)脫嵌鋰機(jī)制的Li多相合金負(fù)極

弱溶劑化少層碳界面實(shí)現(xiàn)硬碳負(fù)極的高首效和穩(wěn)定循環(huán)

全固態(tài)鋰金屬電池負(fù)極界面設(shè)計(jì)





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論