 基于拓撲反應法制備高質量鐵基超導薄膜
基于拓撲反應法制備高質量鐵基超導薄膜
一、薛其坤團隊:基于拓撲反應法制備高質量鐵基超導薄膜
背景介紹
制備高質量的鐵基超導體并進行系統的物性表征是深入認識高溫超導機理的重要途經。在FeCh(Ch=S, Se, Te)基超導體中,單層FeSexTe1-x/SrTiO3(0< x ≤ 1) 體系因其顯著增強的超導電性和非平庸的拓撲性質而受到廣泛關注。掃描隧道顯微鏡和角分辨光電子能譜等實驗手段測得單層FeSexTe1-x的超導能隙閉合溫度在50 ~ 83 K之間,遠高于電輸運測得的小于30 K的零電阻轉變溫度。
能隙閉合溫度和零電阻轉變溫度之差異與薄膜的不均勻性存在密切聯系。單層FeSe通常含有鐵空位造成的密集的線缺陷,其超導能隙在線缺陷附近及線缺陷所包圍的疇中受到顯著抑制。單層FeSexTe1-x存在納米尺度化學相分離,這可能會導致局域的磁有序。
這種樣品的不均勻性既阻礙了高溫超導機理的研究,又限制了單層FeSexTe1-x在量子計算相關研究方面的進一步探索。因此,優化單層FeSexTe1-x超導薄膜質量、提升其空間均勻性對基礎研究和材料應用均具有重要意義。
相比于FeCh基超導體,FePn (Pn = P, As)基超導體具有更為龐大的家族體系(包括由Li、Ba/Sr、LaO和SmO等插層構成“111”,“122”和“1111”等體系)和更為豐富的物理特性(如磁有序結構等)和調控性。特別是,具有鐵基塊體材料最高超導轉變溫度55K的SmOFFeAs列于其中。
四方相結構的FePn為電荷不飽和態,實驗上很難獲得FePn超導表面。開展對FePn超導層的直接表征和調控研究具有與CuO2超導層研究相當的重要性和挑戰性。獲得四方相FePn層是開展相關表征和調控研究的關鍵性環節,對探索FePn基超導材料的配對機制以及提高FePn基材料的超導轉變溫度均具有重要意義。
成果簡介
薛其坤團隊通過拓撲反應法成功制備了系列的高質量單層FeSexTe1-x (0 < x ≤ 1)超導薄膜。研究團隊首先利用分子束外延技術在SrTiO3(001)襯底上生長單疇的單層FeTe薄膜,然后將FeTe薄膜置于Se氣氛下退火。Se的電負性強于Te,在適當條件下,Se可取代薄膜中的Te而形成單層FeSexTe1-x薄膜。精準調控襯底溫度、Se束流及反應時間可實現Se化學配比x在0-1全范圍精準控制。團隊利用掃描隧道顯微鏡對單層FeSexTe1-x的電子態和表面形貌進行了原位表征,測得其超導能隙高達15 meV,并且Te和Se原子隨機分布達到原子尺度化學均一。
特別地,與Fe-Se共沉積方法獲得的單層FeSe相比,本方法制備的單層FeSe的線缺陷在數量和尺寸上均大幅減小,超導相干峰的強度顯著增強。這些結果都表明拓撲反應法制備的FeSexTe1-x單層膜在納米尺度上具有極佳的均勻性,為探究配對對稱性、贗能隙等尚存爭議的物理問題提供了研究載體。這項工作為合成和尋找亞穩相超導薄膜提供了研究范式。詳見https://doi.org/10.1007/s12274-022-4718-3。
沿此途徑,薛其坤團隊通過多層FeTe薄膜與As原子的拓撲反應成功制備出了具有四方相結構的FeAs層,并在低溫沉積鉀原子后獲得了KxFe2As2薄膜。團隊利用掃描隧道顯微鏡對FeAs層和KxFe2As2薄膜的表面形貌和電子態進行了原位表征,發現FeAs層具有重構,而KxFe2As2膜具有沿重構對角線方向、周期為的條紋結構和~11 meV的超導能隙。
此外,輸運測量結果顯示這樣制備的KxFe2As2/FeTe/STO樣品的超導轉變溫度可達~ 10 K,相比體相KxFe2As2的3.8 K提高了一倍。由此,團隊獲得了界面增強超導特性的又一實例。更為重要的是,利用拓撲反應法成功制備具有四方結構的FeAs層為鐵基超導薄膜的制備和調控提供了一個新途徑,為探究鐵基超導體的配對對稱性等超導機制提供了研究載體。詳見https://doi.org/10.1007/s12274-022-4956-4。
圖文導讀
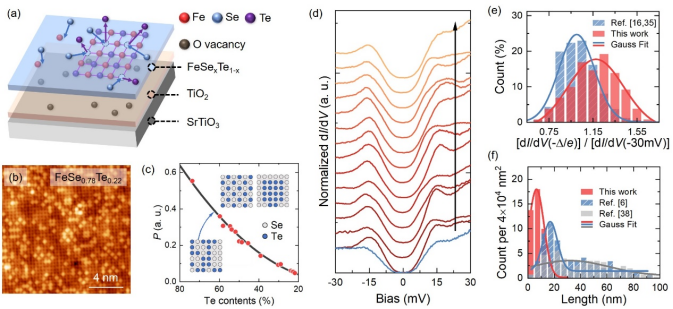
圖1. (a)單層FeTe的硒化反應示意圖。(b) 拓撲反應法制備的單層FeSe0.78Te0.22的原子分辨圖。(c) 不同Te含量樣品的參量P的值(紅色圓點),其中P為最近鄰Te-Te化學鍵數目與陰離子間總化學鍵數目之比,黑色曲線為Te原子隨機分布時P與Te含量的依賴關系。(d) 拓撲反應法制備的單層FeSe的隧道譜,其相干峰高度相較于共蒸發法情形(藍色)有顯著提升。(e) 相干峰高度統計及與共沉積結果對比。(f) 線缺陷數量和長度統計及與共沉積結果對比。
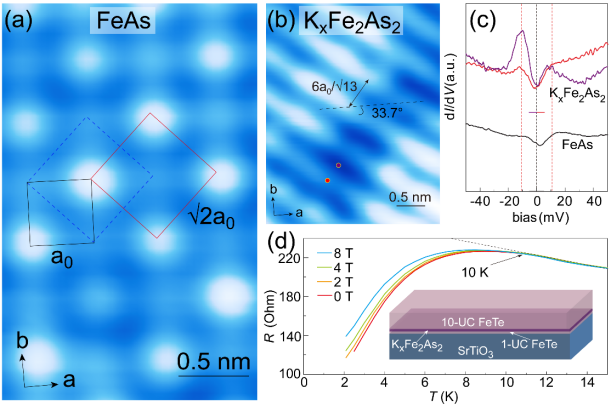
圖2. (a) 拓撲反應法制備的FeAs層的原子分辨圖顯示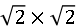 重構。(b) KxFe2As2表面形貌圖顯示沿
重構。(b) KxFe2As2表面形貌圖顯示沿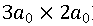 重構對角線方向、周期為
重構對角線方向、周期為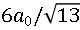 的條紋相。(c) FeAs和KxFe2As2表面的掃描隧道譜線。KxFe2As2表面的掃描隧道譜線顯示± 11 meV處相干峰強度隨條紋結構調制變化。(d) KxFe2As2/FeTe/STO樣品的結構示意圖和R-T測試結果。
的條紋相。(c) FeAs和KxFe2As2表面的掃描隧道譜線。KxFe2As2表面的掃描隧道譜線顯示± 11 meV處相干峰強度隨條紋結構調制變化。(d) KxFe2As2/FeTe/STO樣品的結構示意圖和R-T測試結果。
二、北航賈彬彬/北理工陳卓老師:促進電化學CO2還原的非對稱Ni-P1N3活性中心的合理設計
背景介紹
利用可持續能源發電將二氧化碳(CO2)催化轉化為增值產品正受到越來越多的關注。單位點催化劑(SSC)由于其高活性中心和高原子利用率,在電催化CO2還原反應(CO2RR)中具有優異的性能。從實驗和理論層面來看,貴金屬SSC(如Au、Ag和Pd)對CO2RR寄予厚望。但其稀有性和高昂的價格限制了其實際應用。同時,據報道,Fe、Co、Ni、Mn和其他非貴過渡金屬(TM)SSC在CO2RR中也顯示出高催化活性。
對于蓬勃發展的碳基TM SSC,提出金屬氮(M-Nx)位點可能是有利于CO2吸附和還原反應的活性中心。與中心原子配位的摻雜N的數量和類型的調整將導致過渡金屬電子排列的差異,這將影響CO2RR中間產物的吸附和解吸。否則,還可以通過用外來的元素替換部分N原子來改變配位微環境,例如磷(2.19),與N(3.04)和C(2.55)相比,磷具有更大的半徑和更低的電負性。P的加入改變了原始M-N鍵的長度和中心金屬原子的正電荷。
研究方法
通過熱解的MOF框架構建了界面調控的P、N雙配位的碳基負載的鎳單位點催化劑(表示為Ni-PxNy,x=1,2和y=3,2),應用于電催化CO2還原反應(CO2RR)中。
成果簡介
引入同步輻射XAS測試確定Ni中心原子的局域配位結構。實驗和理論結果表明,Ni-PxNy單分散活性位點是高催化活性的根源。其中Ni-P1N3上的CO電流密度顯著高于未經P改性的Ni-N4催化劑,在-0.65至-0.95 V(vs RHE)的寬電位范圍內具最高FECO,為85.0-98.0%,同時反應20小時后仍能保持初始FECO的94%。實驗和理論結果表明,Ni-P1N3催化劑具有與COOH*和CO*關鍵反應中間體適度的結合強度,因而具有更高的催化CO2還原活性。
圖文導讀
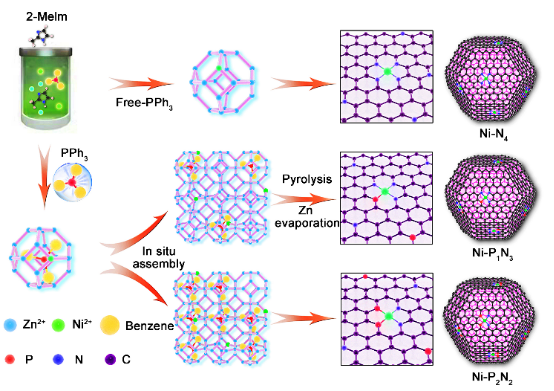
圖1. Ni-PxNy樣品的制備流程示意圖
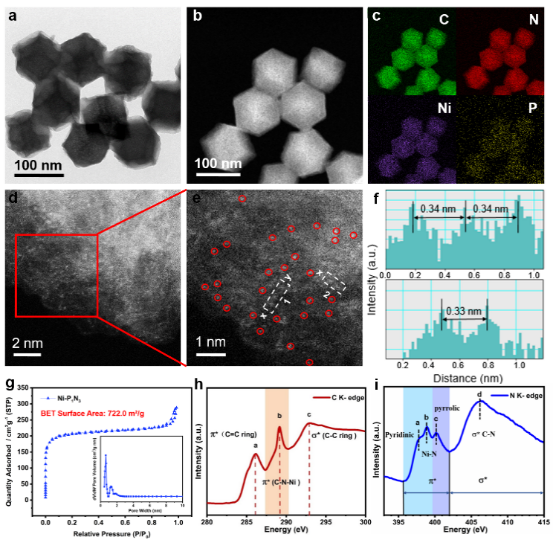
圖2. Ni-P1N3的(a)TEM圖像;(b)STEM圖像和(c)相應的EDS圖像;(d)和(e)HAADF-STEM及其放大圖像;(f)沿e中X-Y線的強度分布;(g)BET表面積和孔徑分布;(h)Ni-P1N3的 C K-邊XANES光譜;(i)N K-邊XANES光譜 圖2c中的圖像上彩色亮點證實了樣品中Ni、P、N和C物種的存在,表明Ni、P和N元素成功摻入且均勻分布在整個碳基結構中。
圖2d、2e上獨立分散的白色亮點為鎳金屬原子,整個碳基材料上并未觀察到鎳金屬納米顆粒的存在,表明Ni金屬原子呈高度單分散狀態。測量圖2e上鎳金屬白色亮點間的距離,沿X-Y的相應強度分布表明(圖2f),單個Ni原子之間的間距至少為0.33 nm,明顯大于單個Ni原子的有效半徑(124.6 pm = 0.125 nm),進一步驗證了Ni物質在制備的Ni-P1N3中的孤立特性。
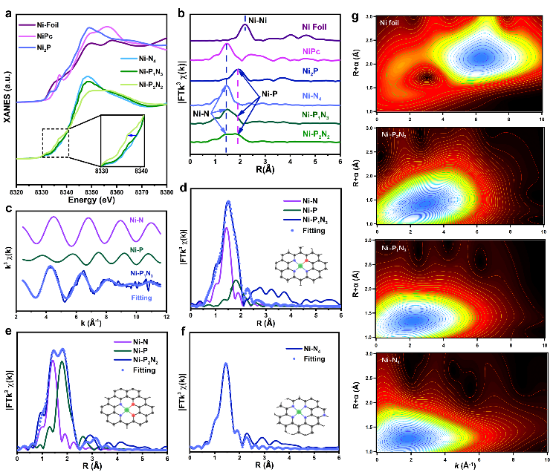
圖3. Ni-PxNy及標準樣品的Ni K邊(a)XANES和(b)EXAFS光譜;Ni-P1N3的(c)k空間和(d)FT-EXAFS擬合曲線;(e)Ni-P2N2和(f)Ni-N4的FT-EXAFS擬合曲線;(g)Ni箔、Ni-P2N2、Ni-P1N3和Ni-N4的WT-EXAF圖 基于同步輻射的XAS測量深入探究原子級Ni SSCs的界面結構。
Ni物種的平均氧化態可以通過Ni K邊的吸收閾值描述。在XANES曲線中(圖3a),Ni-P1N3的位置在Ni-N4和Ni-P2N2之間,表明Ni的氧化態在二者之間。通過對比Ni SSCs樣品與標準樣品的傅里葉變換(FT)EXAFS光譜,探究Ni-P1N3、Ni-N4和Ni-P2N2的界面結構,如圖3b所示。Ni-P1N3樣品在1.44 ?處有一個明顯的FT峰,對比僅含有Ni-N配位的NiPc可知,該峰主要來自Ni-N配位的散射;在1.89 ?處檢測存在一個肩峰,與NiP2的FT-EXAFS光譜進行比較,表明該信號來自Ni-P散射,證實了Ni-P鍵的形成。
此外,與Ni箔相比,在Ni-P1N3、Ni-N4和Ni-P2N2譜圖上均未觀察到源自Ni-Ni的散射峰,證實催化劑上不含有Ni金屬小顆粒,Ni主要以單個原子的形式存在于碳基材料上,同時與N和P原子形成配位結構。進行小波變換處理(圖3 g)進一步驗證Ni的原子色散,與Ni箔相比,Ni-P1N3、Ni-N4和Ni-P2N2譜圖均未在對應位置出現波峰。對Ni-P1N3進行EXAFS擬合定量提取Ni K邊結構參數,擬合結果如圖3c-f所示。
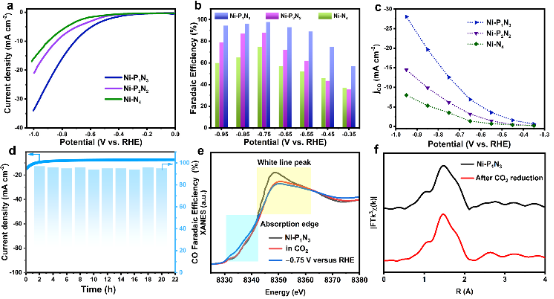
圖4. Ni-PxNy及標準樣品的催化性能和原位XAFS表征。 我們使用三電極H電池評估了CO2飽和0.5 M KHCO3溶液中的CO2RR性能。在線性掃描伏安法(LSV)曲線中(圖4a),Ni-P1N3在-1.0 V時的電流密度為33.98 mA cm-2,而RHE的電流密度遠大于比較的Ni-P2N2(21.82 mA cm-2)和Ni-N4(16.97 mA cm-2)。
此外,當在Ar飽和電解質中測量Ni-P1N3時,圖S19中檢測到電流密度顯著降低,起始電位增加,表明活性來源于CO2RR。通過氣相色譜法和核磁共振波譜法對產物進行了測量,表明在CO2RR過程中僅生成CO和H2。圖4b顯示了Ni SSC的CO法拉第效率(FECO)。其中,Ni-P1N3在整個電位區間內超過Ni-P2N2和Ni-N4,在-0.75 V時達到98%的最大FECO。此外,圖4c中顯示了CO電流密度(jCO)與外加電位的關系。在-0.75 V的電位下,Ni-P1N3顯示出14.30 cm–2的高電流密度,是Ni-P2N2和Ni-N4的2.1倍和3.6倍。
此外,Ni-P1N3也表現出良好的穩定性,在電解20 h后,相對于初始FECO,其保持94%(圖4d)。 為了深入研究CO2RR過程中催化劑的演化,對反應前后的催化劑進行了原位X射線吸收光譜測試。近邊光譜(圖4e)顯示,反應條件下Ni-P1N3的吸收邊緣向較低能量轉移,表明Ni的價態在反應過程中略有下降。對反應前后的結構數據進行對比(圖4f),反應后催化劑的結構沒有發生明顯變化,仍保留著Ni-P1N3四配位的界面結構,具有一定的結構穩定性。
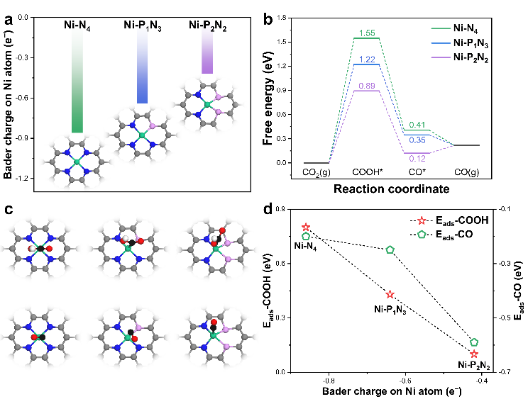
圖5. (a)Ni-N4、Ni-P1N3和Ni-P2N2催化劑的原子結構以及中心Ni原子上的Bader電荷計算值;(b)CO2RR自由能圖;(c)反應中間體的原子結構示意圖(d)COOH*和CO*在中心Ni原子的吸附能(Eads) 為分析Ni-N4、Ni-P1N3和Ni-P2N2催化劑間性能差異的來源,首先計算不同原子界面結構中Ni位點到碳基底的Bader電荷轉移數,如圖5a所示,隨著Ni-N4結構中N原子逐漸被P原子取代,從Ni活性中心轉移到碳基底的電子數量逐漸減少。因此,具有P配位的Ni活性位點保留了更多的電子,這可能會大大增強與吸附物的相互作用。
計算分析Ni-PxNy上CO2RR生成CO的自由能差異,如圖5b。正如之前研究所示,在CO2電催化生成CO通常由兩個步驟決定,即COOH*中間體的形成和CO*的解吸。因此,理想的CO2RR-to-CO催化劑應與COOH*形成強結合,但同時與CO*結合較弱。然而,目前的工作中COOH*和CO*之間存在線性比例關系,為了設計高效的催化劑,規避反應中間體之間比例關系對于設計高效催化劑是必要的。
圖5b的例子中,在Ni-N4催化劑中添加P原子后,COOH*形成自由能從1.55 eV(Ni-N4)逐漸下降到1.22 eV(Ni-P1N3),然后下降到0.89 eV(Ni-P2N2),表明COOH*中間體的結合強度隨著P原子的摻入逐漸增強。但伴隨著對COOH*中間體吸附結合的增強,Ni活性位點上對CO*結合強度也隨之提升,不利于CO的解吸,并最終限制了CO2RR的催化性能。
反應中間體增強的結合強度與Ni活性位點轉移到基底的電荷量有關(圖5d)。反應中間體(即COOH*和CO*)的原子結構及其吸附示意圖(如圖5c)。Ni-N4、Ni-P1N3和Ni-P2N2催化劑上COOH*結合能以相同的幅度增加(0.33 eV),而Ni-P2N2催化劑對CO的吸附強度顯著增加,并從Ni-N4和Ni-P1N3的物理吸附轉變為化學吸附,表明CO在Ni-P2N2催化劑上的解吸過程緩慢。
因此,在Ni-N4上,CO2RR的性能受到COOH*產生的限制,而Ni-P2N2的性能則更多受CO*脫附的影響。顯然,在這種情況下,CO分子穩定地吸附在Ni原子上,不利于CO2RR過程的進行。在這方面,具有中等COOH*和CO*中間體結合強度的Ni-P1N3催化劑則是CO2RR轉化為CO的最理想催化劑,COOH*的形成和CO*解吸都不會過于限制整個反應過程的限制。DFT結果與實驗測試結果一致,Ni-P1N3表現出比Ni-N4和Ni-P2N2增強的CO2RR活性。
三、張袁斌課題組:具有柔性篩分效應的新型陰離子柱撐MOF用于從CO2和CnH4中高效捕獲C2H2
背景介紹
乙炔(C2H2)是工業上生產各種必需化學品和聚合物的主要原料。它是由天然氣的部分燃燒或碳氫化合物的裂解產生的,其中二氧化碳(CO2)和其他C1 -C2碳氫化合物會與其共存,需要去除以生產高純度的C2H2。目前,采用能耗較高的低溫蒸餾和溶劑萃取法從其他氣體中提取C2H2。由于沸點相近,這些方法的效率低、能耗高且對環境有污染。
因此,基于多孔固體吸附劑的物理分離方法由于較低的成本和能耗而引起了人們的關注。然而,這些氣體分子在分子大小(動力學直徑:C2H2和CO2均為3.3?,CH4為3.8?,C2H4為4.2?)和物理性質方面的相似性也加大了分離的難度。金屬有機框架(MOFs)以其強大的結構可預測性和對孔隙大小/形狀和功能的可調性而被廣泛關注。
研究方法
慢擴散方法
ZNU-4的合成方法:在一個5mL的玻璃管中,在最下層加入1mL含有(NH4)2TiF6(1mg)和Cu(NO3)2?3H2O(1mg)的水溶液,中間加入2mL MeCN/H2O(1:1)的緩沖溶液,最上層加入1 mL DIB的MeCN溶液。然后將玻璃管靜置放置在室溫下,過幾天會在中間層看到有藍色塊狀晶體產生。
ZNU-5的合成方法:在一個5mL的玻璃管中,在最下層加入1mL含有(NH4)2TiF6(1mg)和Cu(NO3)2·3H2O(1mg)的水溶液,中間加入3mL MeOH/H2O(1:1)的緩沖溶液,最上層加入1 mL DIB的MeOH溶液。然后將玻璃管靜置放置在室溫下,過幾天會在中間層看到有紫色針狀晶體產生。
成果簡介
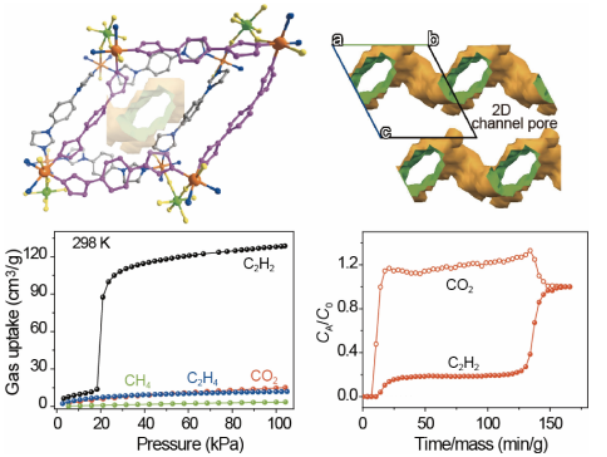
基于以上思考,我們報道了一種新的TiF62?陰離子(TIFSIX)柱撐的金屬有機框架材料ZNU-5 (ZNU =浙江師范大學),該材料可通過門開產生的分子篩分效應,以低吸附熱選擇性捕獲C2H2。ZNU-5在1.0 bar和298 K時吸附大量的C2H2(128.6 cm3 /g), 但對CO2、CH4和C2H4的吸附微乎其微。如此高的吸附量在分子篩分的MOFs中還沒有實現過。
穿透實驗進一步證實了該材料可從多組分混合氣體中高選擇性地捕獲C2H2。ZNU-5可分別從等摩爾C2H2/CO2、C2H2/CO2/CH4和C2H2/CO2/CH4/C2H4混合物中捕獲3.3、2.8和2.2 mmol/g的C2H2。此外,2.6、2.0和1.5 mmol/g,且純度> 98%的C2H2可以從解吸過程中回收。ZNU-5結合高的吸附容量、低吸附熱、良好的可回收性,具有良好的C2H2純化前景。
圖文導讀
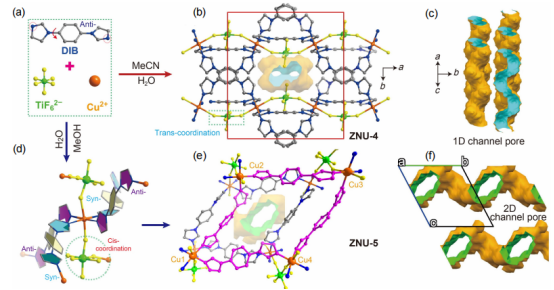
圖 1 (a) ZNU-4和ZNU-5的結構組成單元。(b) ZNU-4的多孔結構。(c) ZNU-4的孔隙。(d) ZNU-5中的配位模式。(e) ZNU-5的多孔結構。(f) ZNU-5的孔隙。孔隙由半徑為1.2 ? 的探針進行測試。
要點:圖1a是ZNU-4和ZNU-5的組成單元,兩種材料合成方法的差異在于慢擴散過程中使用溶劑的不同。ZNU-4是由MeCN/H2O溶劑參與慢擴散形成的,而ZNU-5是由MeOH/H2O參與的慢擴散所獲得的。圖b是ZNU-4的框架結構,在ZNU-4中每個Cu(II)離子與來自四個不同DIB配體的四個咪唑氮原子和來自兩個TiF62-基團的兩個氟原子相連。
其中,DIB配體為anti-構型,而TiF62-通過trans構型配位。6個Cu2+和6個DIB配體產生一個非常扭曲的環。每個單元中都包含兩個相鄰的狹窄的一維通道,表面裝飾著氟原子。圖d是ZNU-5的框架結構,其中Cu(II)為八面體配位,連接四個DIB配體(一半采取syn-構型,一半采取anti-構型)和兩個TIFSIX陰離子(采取cis-構型),形成具有pcu拓撲結構的非互穿框架。與ZNU-4不同,ZNU-5具有二維的孔隙通道。
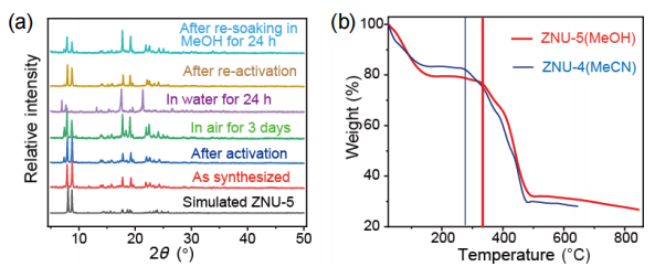
圖2(a)不同條件下ZNU-5的PXRD圖。(b)ZNU-4和ZNU-5的熱重分析曲線。 要點:從PXRD的測試結果可以看出制備的ZNU-5具有很好的相純度,且在活化后和空氣中放置三天,材料依舊穩定。但將該材料浸泡在水中24h后,得到了新的相,但將水中浸泡的樣品再次活化或浸泡甲醇后,依舊可以回到合成的相狀態,表明其相的轉變具有可逆性。從熱重的測試結果可以看出,ZNU-4的熱穩定性在290℃,ZNU-5較ZNU-4的熱穩定性高,為340℃。
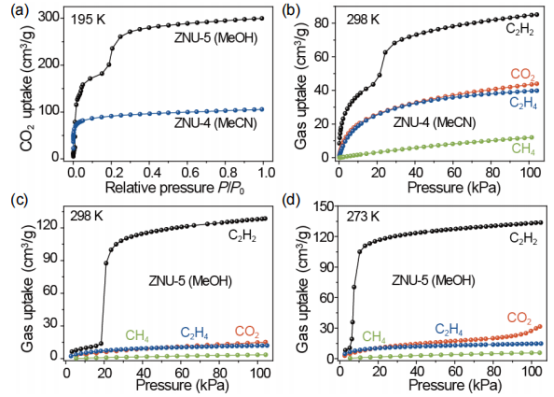
圖3 (a) ZNU-4和ZNU-5在195 K時的CO2吸附等溫線。(b)在298 K時,ZNU-4中C2H2、CO2、C2H4和CH4的吸附等溫線。(c,d)在298K和273 K時,ZNU-5中C2H2、CO2、C2H4和CH4的吸附等溫線。 要點:圖a為195K下ZNU-4和ZNU-5的吸附曲線。測試結果顯示ZNU-4的吸附呈現I型吸附等溫線,在100 kPa時最大吸附量為105.7 cm3 g-1,BET表面積為358.6 m2·g-1。
與ZNU-4不同,ZNU-5的CO2吸附過程可分為兩個階段,第一步和第二步的CO2吸附量分別為200.9 cm3g-1(15 kPa)和299.8 cm3g-1(100 kPa),BET表面積為751.5 m2·g-1。計算得到的孔徑為5.2 ?。氣體吸附等溫線顯示,ZNU-5在298K時對C2H2存在門開效應,而對C2H4、CH4和CO2卻沒有該現象。
其在298 K和100 kPa時表現出較高的C2H2吸附量為128.6 cm3g-1,比ZNU-4(85.1 cm3·g-1)的吸附量高51.1%。與此同時,在298 K時,當壓力高達100 kPa時,ZNU-5完全阻止了CO2、C2H4和CH4的進入。因此,在100 kPa下較高的C2H2吸附能力,以及較低的CO2、C2H4和CH4吸附,使ZNU-5具有從C2H2/CO2/C2H4/CH4四組分混合物中實現一步純化C2H2的能力。
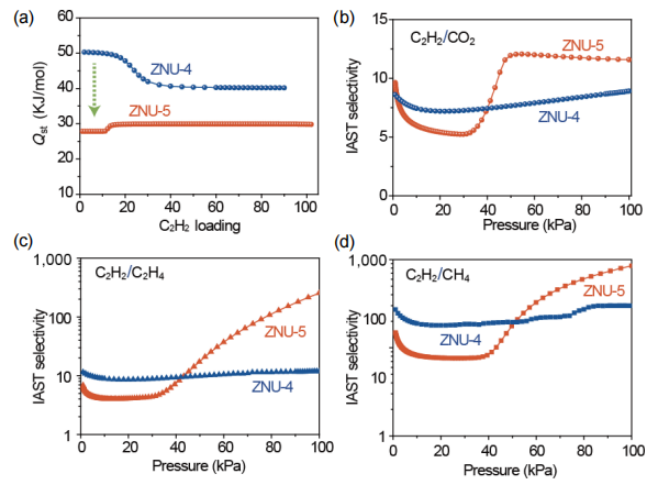
圖4 (a) 在ZNU-4和ZNU-5中C2H2的Qst。(b-d)在298K下,ZNU-4和ZNU-5對等摩爾C2H2/CO2、C2H2/C2H4和C2H2/CH4混合物的IAST選擇性。
要點:圖a為ZNU-4和ZNU-5對C2H2的Qst值。其中,ZNU-5的C2H2Qst值(27.8 kJ·mol-1)遠低于ZNU-4(50.3 kJ·mol-1),說明其再生能耗相對較低。圖b、c和d為ZNU-4和ZNU-5對C2H2/CO2、C2H2/C2H4和C2H2/CH4的IAST選擇性比較。由于ZNU-5中存在對于C2H2的門開效應,使其IAST選擇性較ZNU-4有所提升。ZNU-5在100 kPa時具有較高的選擇性,分別高達12、255和850,優于ZNU-4的選擇性,分別為9、12和300。
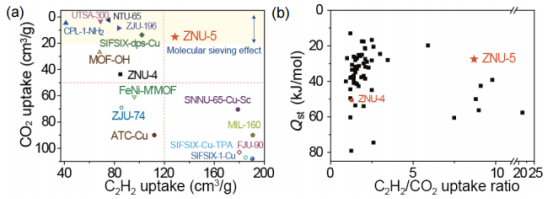
圖5(a) ZNU-4/ZNU-5和其他材料在100 kPa和298K時,C2H2和CO2吸附量的比較。(b) ZNU-4/ZNU-5與其他材料對C2H2的Qst及C2H2/CO2的吸附容量比比較。 要點:通過與其他材料對比,ZNU-5顯示出非常高的C2H2吸附和非常低的CO2吸附,這也反映在圖5b中的C2H2/CO2吸附比中。此外,ZNU-5是極少數具有C2H2/CO2吸收比>8且Qst值< 30 kJ/mol的多孔材料。 ?
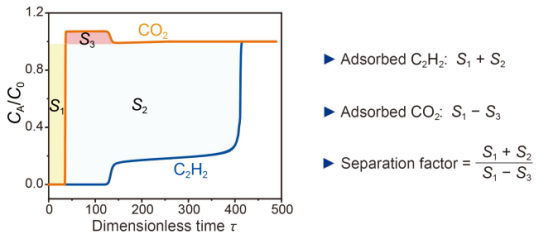
圖6ZNU-5在298 K下對C2H2/CO2(50/50)的突破曲線。
要點:瞬時穿透測試評估了ZNU-5對等摩爾C2H2/CO2(50/50)混合物的分離性能。如圖6所示,ZNU-5對C2H2呈階梯式穿透曲線,這在剛性吸附劑中從未觀察到過。通過對曲線的分析可看出捕獲的C2H2量仍然很大,而CO2量可以忽略不計,從而得到了一個較高的分離系數。此外,C2H2需要解吸過程才能獲得,因此穿透過程中的輕微泄漏不會對C2H2的動態捕獲有很大的影響。
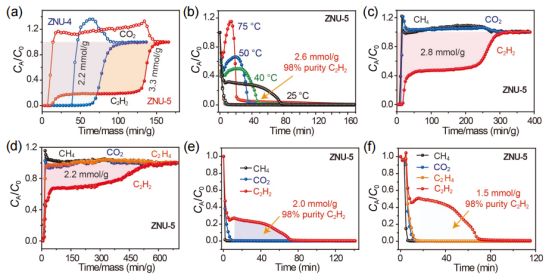
圖7 (a) ZNU-4和ZNU-5的C2H2/CO2 (50/50)穿透曲線。(b)ZNU-5在25℃、40℃、50℃和75℃時的C2H2/CO2 (50/50)解吸曲線。(c)在ZNU-5中,C2H2/CO2/CH4 (33.3/33.3/33.3)穿透曲線。(d)在ZNU-5中, C2H2/CO2/C2H4/CH4(25/25/25/25)分離的穿透曲線。(e)ZNU-5中C2H2 /CO2 /CH4 (33.3/33.3/33.3)分離的解吸曲線。(f)ZNU-5對于C2H2/CO2/C2H4/CH4(25/25/25/25)的解吸曲線。
要點:為了評估實際的分離性能,并確定階梯式穿透現象,進行了實驗穿透測試。實驗結果表明,實驗穿透曲線與模擬結果非常相似。對于等摩爾的C2H2/CO2混合物,ZNU-5可以實現有效的分離,可以獲得3.3 mol·g-1的C2H2,而ZNU-4吸附C2H2的量僅為2.2 mol·g-1。隨后,研究了不同的解吸溫度對再生的影響。從圖7b可以看出,隨著解吸溫度的升高,C2H2的解吸時間逐漸縮短。
在解吸溫度為25℃時,吹出CO2后,可從柱中回收2.6 mol·g-1純度為98%的C2H2。后續的三組分及四組分穿透測試仍然得到了高純度的C2H2,證明了ZNU-5仍具有較好的從三組分和四組分混合物中得到高純C2H2的能力。
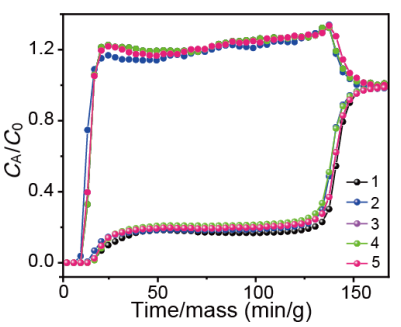
圖8 C2H2/CO2(50/50、v/v)混合物的動態穿透曲線的五次循環。
要點:在五次循環穿透測試中,ZNU-5材料的穿透時間及容量均沒有發生變化。證明該材料是一種很有前途的吸附劑,具有良好的循環穩定性。
四、廣東工業大學易國斌、林霄峰課題組:彈性模量可控的自擦除動態表面圖案助力多重信息存儲與抗篡改應用
背景介紹
近年來,動態表面圖案(DSPs)由于其對編碼表面功能和性質的按需控制而得到了迅速發展,有望應用于光電器件、智能窗戶、防偽,尤其是在信息安全方面。通過將智能材料引入褶皺以響應外部刺激(例如濕度、光、溫度、pH 和拉伸應力),可以制造可控和響應迅速的 DSP,并針對表面形貌和功能進行動態調整。
相比單模式、雙模式,甚至多模式的光學圖案化技術,動態表面圖案可以有效提高信息存儲容量和加密能力。盡管許多研究報告了基于褶皺和熒光的雙模式 DSPs發展,用以加密復雜信息,但由于 DSP 中不可控的褶皺保留時間和相互干擾的皺紋和熒光狀態,應用于供應鏈的防篡改仍然是一個非同尋常的挑戰。
因此,本研究旨在開發和設計一種具有快速響應的新型DSPs,可以控制褶皺保留時間并獨立調節褶皺和熒光狀態中的不同信息。這種新策略可以為產品安全或無墨印刷中的防篡改、高密度和多編碼信息存儲提供一種新方法。
成果簡介
本研究開發了一種基于可控彈性模量的圖案化策略,以制造具有更高信息存儲密度的自擦除動態表面圖案 (S-DSPs)。這些新型 S-DSPs 戰略性地將氨基共聚物(ACO、DMAEMA-co-DMAPAA)與 9-蒽甲醇 (9-AM) 作為表層,聚二甲基硅氧烷 (PDMS) 作為柔性基板,設計成雙層多編碼體系。它可以攜帶幾種不同類型的信息,如褶皺和熒光。
為了賦予皮膚層更寬范圍的彈性模量,我們通過甲基丙烯酸 2-(二甲氨基)乙酯 (DMAEMA) 和 N, N-二甲氨基丙基丙烯酰胺 (DMAPAA) 的自由基聚合合成了相對低分子量的共寡聚物(ACOs),同時皮膚層中9-AM的羥基和ACOs的酰胺基會進行結合形成氫鍵構成超分子交聯網絡。
隨后,雙層系統中壓縮應變和臨界褶皺應變之間的差異可以通過皮膚層的光二聚作用來精確調節,從而使 S-DSPs 產生快速響應(最小< 1 分鐘)和可自擦除的褶皺圖案(3 分鐘~8 天)。此外,熒光圖案可以在不改變褶皺圖案的情況下獨立地被擦除和重新編程,從而開發出具有多種編碼信息存儲能力的 S-DSPs。
同時,具有可控自擦除功能的S-DSP還可用于產品的保質期,極大地提高了供應鏈的防篡改能力。據我們所知,這是控制褶皺保留時間并獨立調節褶皺和熒光狀態中不同信息的DSPs的第一份報告,該策略可以應用于防篡改、高密度和高編碼信息存儲的產品安全、智能顯示器和無墨打印。
圖文導讀
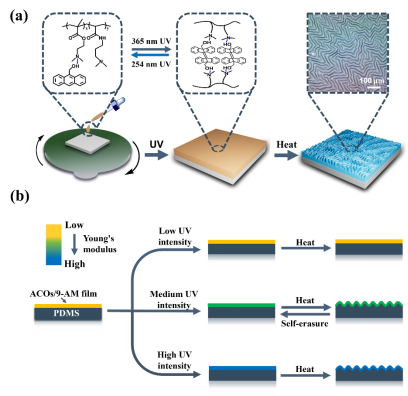
圖1 S-DSPs的調控策略。(a) 制備 DSPs的策略。(b) 三種不同紫外線強度下起皺變化的示意圖。
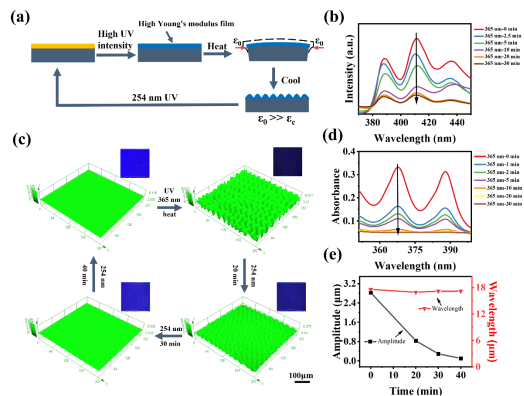
圖 2 褶皺和熒光同時變化的 DSPs。(a) 高強度紫外線照射的起皺動力學。(b) ACOs/9-AM 薄膜在 365 nm 紫外光下不同照射時間的熒光光譜。(c) 樣品在 365 nm 紫外線照射后通過熱處理起皺和在 254 nm 紫外線照射下消除皺紋的 LSCM 圖像。插圖是相應的熒光圖像,比例尺:100 μm。(d) ACOs/9-AM 薄膜在 365 nm 紫外光下不同照射時間的紫外-可見光譜。(e)褶皺擦除過程中波長和幅度的詳細變化。

圖 3 在紫外線照射下通過光掩模形成的褶皺和熒光圖案。(a) 在自然光和紫外光下未處理樣品的圖像。放大圖像顯示樣品表面透明且無褶皺,比例尺:5 mm。(b) 365 nm 紫外線照射下的“蝴蝶”褶皺和相應的熒光圖案照片(紫外線強度:1000 mW·cm-2)。放大的圖像顯示了樣品內部褶皺的無序形狀,比例尺:100 μm。(c) 帶有“條紋”光掩模,在 254 nm 紫外線照射下褶皺消除和熒光增強的圖案。放大的圖像顯示了擦除的細節,比例尺:5 mm。

圖 4 僅有熒光變化的 DSP。(a) 低強度紫外線照射的起皺動力學(紫外線強度:250 mW·cm-2)。(b) 低強度紫外線照射后再高溫加熱處理的圖案變化。比例尺 1:5 mm 比例尺 2:4 mm。(c) Ⅰ在高紫外線強度(紫外線強度:1000 mW·cm-2)的照射下形成褶皺和熒光的“工”圖案;Ⅱ高溫加熱后“工”的熒光圖案消失;Ⅲ低強度紫外線照射后的熒光圖案發生變化。比例尺 1: 5 mm比例尺 2:4 mm。
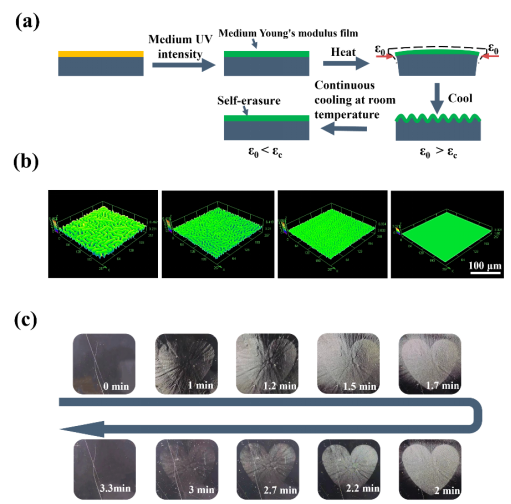
圖 5 具有可自擦除褶皺的 DSPs。(a) 中等強度紫外線照射的起皺動力學(紫外線強度:600 mW·cm?2)。(b) 中等強度 365 nm 紫外線照射下褶皺自擦除的 LSCM 圖案,比例尺:100 μm。(c)隨著時間的推移褶皺自我擦除的光學圖像。
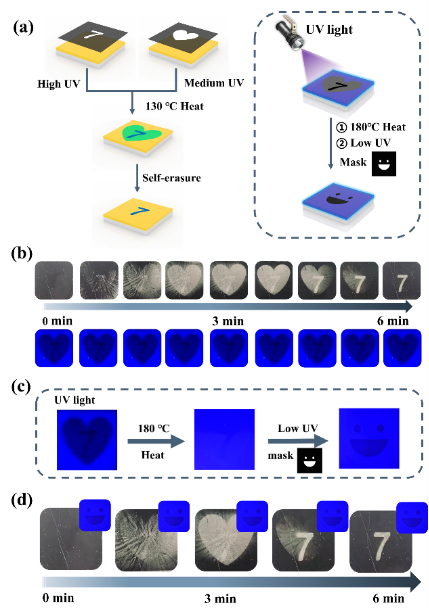
圖 6 用于多編碼信息存儲的 S-DSPs。(a) 多編碼 S-DSPs的設計思路。(b)在自然光下具有褶皺變化的 S-DSPs的光學圖像。(c) 熒光圖案重構的過程。(d) S-DSPs 的褶皺和熒光變化。
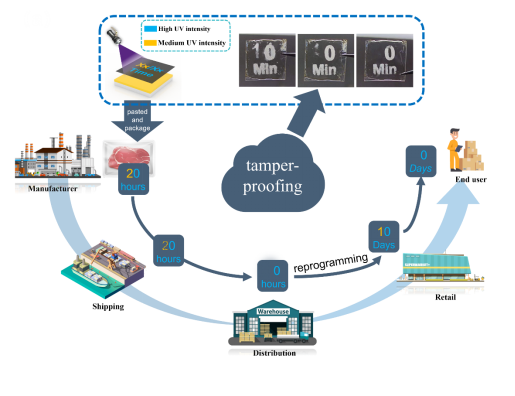
圖 7 S-DSPs 的可控自擦除在供應鏈中防篡改應用。
審核編輯:劉清
-
DFT
+關注
關注
2文章
231瀏覽量
22740 -
ssc
+關注
關注
0文章
24瀏覽量
11218 -
傅里葉變換
+關注
關注
6文章
441瀏覽量
42607
原文標題:Nano Res.:薛其坤/賈彬彬/陳卓等納米合成研究進展
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
芯導科技榮獲上市公司高質量發展大會“科技創新獎”
超導材料的制造工藝 超導材料的分類與比較
產業革命?液相法制備碳化硅襯底實現交付!
立洋光電助力城市照明高質量發展
中興通訊引領5G-A高質量發展新紀元
揭秘高質量點焊機的五大標準:打造焊接性能的基石

科技創新!國產自主三坐標測量機推動產業高質量發展

鈣鈦礦太陽能電池:優化薄膜質量與精準厚度測量

洲明大屏參與中央廣播電視總臺高質量發展“南粵之窗”揭牌儀式

維信諾高質量發展創新大會暨全球合作伙伴大會召開
京東方華燦光電獲評國家級綠色工廠,助力行業高質量發展
北斗芯片產業的高質量發展之路

富捷電子被授予“高質量發展突出貢獻獎”
穩中創新?產業升級?高質量發展 | 聯誠發高質量發展工作推進會議召開

捷易科技出席廣東省韶關市高質量發展大會






 工商網監
工商網監
評論