") 設(shè)計(jì)一款新的二乙二醇二甲醚(DME)基電解液
設(shè)計(jì)一款新的二乙二醇二甲醚(DME)基電解液
【研究背景】
電解液溶劑化結(jié)構(gòu)化學(xué)最近引起了電池領(lǐng)域研究者的極大關(guān)注,其主要原因是傳統(tǒng)認(rèn)為的通過電解液在電極表面分解形成的固體電解質(zhì)界面膜(SEI)并非穩(wěn)定電極及電池性能的唯一主導(dǎo)因素。這一觀點(diǎn)雖然已經(jīng)通過循環(huán)的石墨、合金、金屬負(fù)極以及多數(shù)正極在“電解液交換實(shí)驗(yàn)”中得到證明,但是并不能驗(yàn)證SEI膜在某些方面可能的積極作用。
因此,如何區(qū)分電解液溶劑化結(jié)構(gòu)及其界面模型與SEI對(duì)電極及電池性能的影響仍具挑戰(zhàn)。尤其將成膜添加劑引入電解液中時(shí),該問題變得更加復(fù)雜,因?yàn)樘砑觿┯锌赡苄纬蒘EI,亦有可能改變電解液溶劑化結(jié)構(gòu),然其各自的作用極難以辨別和區(qū)分。
針對(duì)上述問題,本研究以銻(Sb)負(fù)極為例,設(shè)計(jì)了一款新的二乙二醇二甲醚(DME)基電解液,詳細(xì)闡述了溶劑化結(jié)構(gòu)及其衍生的界面模型和SEI對(duì)電極性能的影響,尤其對(duì)添加劑在電解液體相、電極界面以及SEI成膜過程中的作用進(jìn)行了詳盡解析。
近期,中科院長春應(yīng)化所明軍,蘭州大學(xué)張俊麗,以及韓國漢陽大學(xué)Yang-Kook Sun闡明了電解液組分(溶劑、鋰鹽、濃度、添加劑等)從溶劑化結(jié)構(gòu)、界面模型到 SEI 成膜過程中的分子、離子行為以及各自產(chǎn)生的作用,發(fā)現(xiàn)了獨(dú)特的溶劑化結(jié)構(gòu)衍生的界面模型和SEI均是穩(wěn)定Sb負(fù)極的關(guān)鍵。
其中,添加劑(二氟草酸硼酸鋰,LiDFOB)不但能削弱Li+-DME之間的相互作用,而且參與形成的特定的SEI膜能夠有效削弱電極給電子的能力,進(jìn)而提高電解液及電極穩(wěn)定性。
【內(nèi)容表述】
1.研究主旨
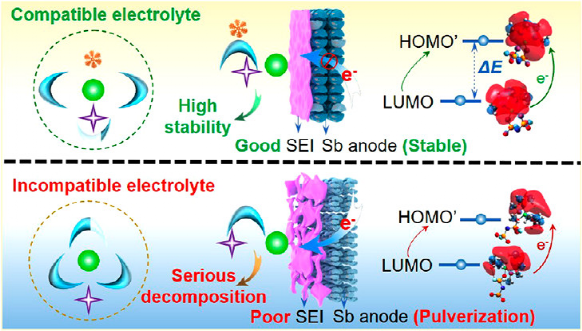
圖1. 溶劑化結(jié)構(gòu)衍生的界面模型和SEI對(duì)電極的影響。
本研究以Sb負(fù)極例,詳細(xì)研究了溶劑化結(jié)構(gòu)衍生的界面模型和SEI對(duì)電極穩(wěn)定性的影響(圖1)。
2.電化學(xué)性能表征
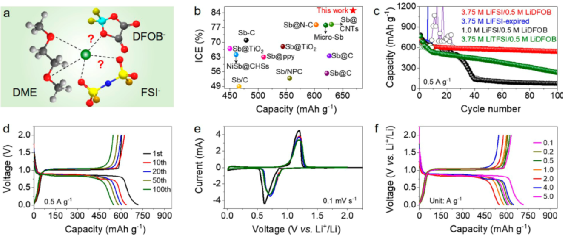
圖2. 微米Sb負(fù)極電化學(xué)性能。
本研究設(shè)計(jì)了一款以LiFSI鋰鹽,DME溶劑,LiDFOB添加劑的電解液(即3.75 M LiFSI/0.5 M LiDFOB in DME),用以穩(wěn)定微米Sb負(fù)極,在該電解液中表現(xiàn)出656 mAh/g的容量及87.5%的首效,優(yōu)于之前報(bào)道的工作(圖2a, b)。0.5 A/g電流密度下,100次循環(huán)后容量保持率88.6%,表現(xiàn)出優(yōu)異的電化學(xué)性能。
相比之下,在沒有LiDFOB添加劑的3.75 M LiFSI in DME電解液中,微米Sb負(fù)極在幾個(gè)循環(huán)后就無法再正常充電。將電解液濃度降低至1.0 M(即1.0 M LiFSI/0.5 M LiDFOB in DME)時(shí),微米Sb負(fù)極容量急劇下降,約40次循環(huán)后容量?jī)H剩~130 mAh/g。
此外,使用LiTFSI鋰鹽(3.75 M LiTFSI/0.5 M LiDFOB in DME),Sb負(fù)極容量也大幅衰減,容量由初始的626 mAh/g經(jīng)100次循環(huán)后降至229 mAh/g。以上結(jié)果表明,電解液的組分(如鋰鹽、溶劑、濃度、添加劑)均會(huì)影響微米Sb負(fù)極性能。
3.電極及SEI膜的表征
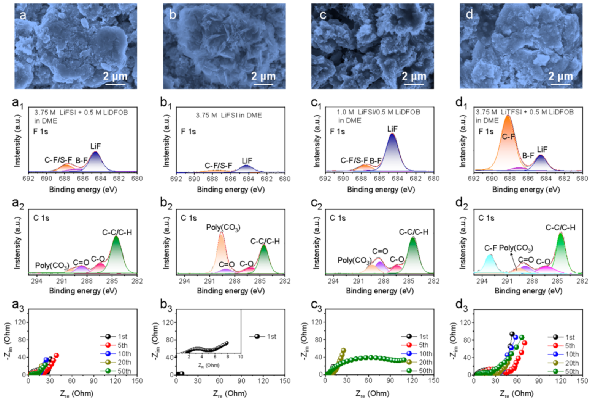
圖3. 電極及SEI膜的表征。
為確定不同電解液對(duì)微米Sb負(fù)極影響的原因,研究者對(duì)循環(huán)后的微米Sb負(fù)極進(jìn)行了相關(guān)表征(圖3)。
結(jié)果表明,(1)3.75 M LiFSI/0.5 M LiDFOB in DME電解液中循環(huán)后的微米Sb負(fù)極表面的SEI比較光滑,且電極粉化程度最小,證明了高濃度和添加劑是維持微米Sb負(fù)極穩(wěn)定的必要條件;(2)從F1s和C1s譜圖可以看出,3.75 M LiFSI/0.5 M LiDFOB in DME電解液中,微米Sb負(fù)極表面形成了一層以LiF為主導(dǎo)(LiF/含F(xiàn)有機(jī)化合物,2.74)且C含量少的SEI,表明在該電解液中,DFOB-的分解量有限,生成的SEI穩(wěn)定,能有效抑制溶劑的分解;(3)3.75 M LiFSI/0.5 M LiDFOB in DME電解液中循環(huán)后微米Sb負(fù)極的電荷轉(zhuǎn)移電阻僅為4.11 Ω,這有利于Li+在電極中的擴(kuò)散。
4.SEI膜作用分析
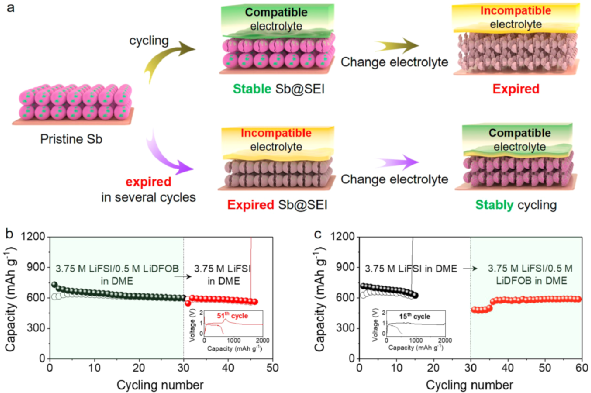
圖4. 電解液交換實(shí)驗(yàn)。
盡管圖3中對(duì)SEI的表征結(jié)果看似可以解釋圖2c中微米Sb負(fù)極不同的電化學(xué)性能,但對(duì)SEI的分析仍不能全面地解釋電解液交換實(shí)驗(yàn)中電極性能的不同。具體的交換實(shí)驗(yàn)如圖4a和b所示。首先,讓微米Sb負(fù)極在兼容電解液(即3.75 M LiFSI/0.5 M LiDFOB in DME)中循環(huán)30次,以此在電極表面形成穩(wěn)定的SEI(Sb@SEI);然后,拆開電池,使用Sb@SEI負(fù)極重新組裝電池,使用不兼容電解液(即3.75 M LiFSI in DME)繼續(xù)循環(huán)。
從圖4c可以發(fā)現(xiàn),在不兼容電解液中,Sb@SEI負(fù)極能夠維持14次循環(huán),在第15次循環(huán)過程中失效。以上結(jié)果表明,如果電解液不能兼容微米Sb負(fù)極,即便在兼容電解液中生成的SEI可以起到保護(hù)微米Sb負(fù)極的作用,但作用有限,不能在長循環(huán)中持續(xù)穩(wěn)定負(fù)極。
相反地,首先讓微米Sb負(fù)極在不兼容電解液(即3.75 M LiFSI in DME)循環(huán),直到其不能正常工作,得到在差的SEI包覆的微米Sb負(fù)極(Sb@SEI);然后,拆開電池,使用Sb@SEI負(fù)極重新組裝電池,使用兼容電解液(即3.75 M/0.5 M LiDFOB LiFSI in DME)繼續(xù)循環(huán)。從圖4d可以觀察到,Sb@SEI負(fù)極經(jīng)過5次循環(huán)后可恢復(fù)初始的容量,并能維持良好的循環(huán)穩(wěn)定性。以上結(jié)果表明,微米Sb負(fù)極失效的原因可能是電解液的不兼容。
5.體相電解液性質(zhì)表征
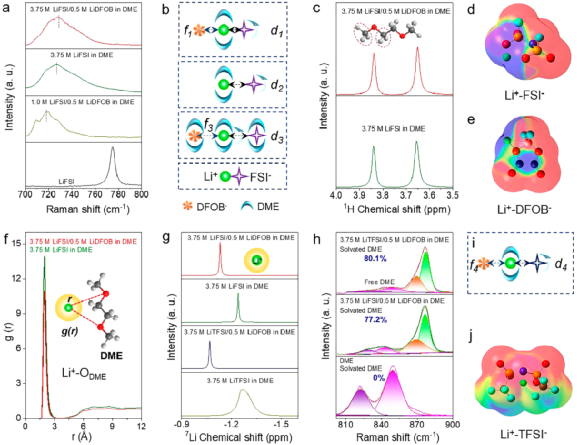
圖5. 電解液表征。
因此,本研究將重點(diǎn)從分子層面上構(gòu)連電解液組分與電池性能之間的關(guān)系。通過拉曼光譜、液相核磁共振譜等測(cè)試分析了不同電解液中的Li+、溶劑、添加劑之間的相互作用,以明晰不同電解液中Li+溶劑化結(jié)構(gòu)的差異(圖5)。拉曼結(jié)果表明,通過提高鋰鹽濃度(如將電解液濃度從1.0 M提高到3.75 M)或引入LiDFOB添加劑可以顯著降低S-N-S的紅移程度。
這主要是因?yàn)殡S著鋰鹽濃度的提高或添加劑的引入,參與溶劑化的DME數(shù)量逐漸不足。換句話就是,紅移程度越小,Li+-FSI-的相互作用越強(qiáng),即3.75 M LiFSI/0.5 M LiDFOB in DME電解液中Li+-FSI-的相互作用最強(qiáng)(圖5a, b)。在1H核磁共振譜中,DME溶劑分子中的-CH2/-CH3基團(tuán)的化學(xué)位移進(jìn)一步證明了引入LiDFOB添加劑能增強(qiáng)Li+-FSI-的相互作用(圖5c)。當(dāng)LiDFOB引入3.75 M LiFSI in DME電解液中,1H核磁共振譜中對(duì)DME中-CH2/-CH3基團(tuán)的屏蔽作用會(huì)增大。
這主要是因?yàn)長iDFOB也需要DME溶劑參與解離,導(dǎo)致溶劑化結(jié)構(gòu)中DME溶劑分子數(shù)量不夠,在一定程度上減弱了Li+-DME的相互作用。這一觀點(diǎn)通過徑向分布函數(shù)計(jì)算結(jié)果得到了進(jìn)一步證明(圖5f)。此外,通過7Li核磁共振譜進(jìn)一步證實(shí)了LiDFOB添加劑對(duì)Li+溶劑化結(jié)構(gòu)的影響。
如圖5g所示,將LiDFOB引入3.75 M LiFSI in DME電解液中后,7Li核磁共振譜的化學(xué)位移向低場(chǎng)偏移表明溶劑對(duì)Li+的屏蔽作用降低,這主要是由于DME溶劑數(shù)量不足和DFOB-在Li+周圍頻率出現(xiàn)增大引起的。這些有力的證據(jù)表明,LiDFOB除了眾所周知的成膜功能外,還能參與Li+溶劑化結(jié)構(gòu)。
6.溶劑化結(jié)構(gòu)及其界面模型
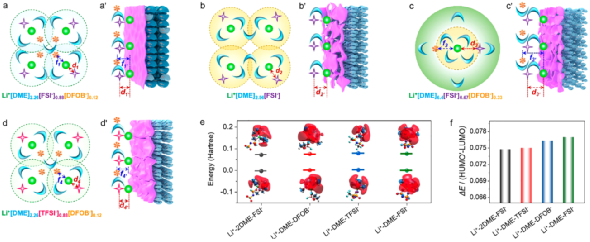
圖6. Li+溶劑化結(jié)構(gòu)及界面模型。
根據(jù)Li+在不同電解液中與溶劑、陰離子之間的相互作用,建立了Li+溶劑化結(jié)構(gòu)及其衍生的界面模型。其中,電解液以其結(jié)構(gòu)化單元Li+[solvent]x[anion]來表示。此外FSI-和Li+之間的相對(duì)距離(即d')以及DFOB-和Li+之間的相對(duì)距離(即f')用于定性描述Li+去溶劑化過程中微米Sb負(fù)極表面Li+-FSI-和Li+-DFOB-相互作用強(qiáng)度,距離越長,則代表相互作用越弱。
此外,用最低的未占用分子軌道(LUMO)和獲得一個(gè)額外的電子時(shí),相應(yīng)軌道變?yōu)镠OMO'的能量差(ΔE)來評(píng)估電解液的穩(wěn)定性。其中,較小的能量差異表示軌道之間的相似性,導(dǎo)致電子傳輸所需要的能量越低,說明了Li+-solvent-anion復(fù)合物穩(wěn)定性越弱,即電解液也不穩(wěn)定。
在3.75 M LiFSI/0.5 M LiDFOB in DME (Li+[DME]2.26[FSI-]0.88[DFOB-]0.12)電解液中,由于DME溶劑不足,不足以將Li+、FSI-和DFOB-充分溶劑化,Li+第一溶劑化層會(huì)共享溶劑而相互重疊。由于Li+-FSI-和Li+-DFOB-的相互作用較Li+-DME強(qiáng),F(xiàn)SI-和DFOB-會(huì)出現(xiàn)在第一溶劑化層中的Li+周圍,削弱Li+與DME之間的相互作用,進(jìn)而降低DME溶劑分子的極化。在去溶劑化過程中,可以在微米Sb負(fù)極表面形成以陰離子為主的電解液分解,有效抑制了溶劑的分解,提高了電解液及微米Sb負(fù)極的穩(wěn)定性(圖6a)。
此外,理論模擬得到,Li+-DME-DFOB-配合物的ΔE(0.0750 Hartte)略小于Li+-DME-FSI-配合物的(0.0770 Hartte)(圖6f)。這說明,Li+-DME-DFOB-配合物更容易接收電極上的電子,并在電極表面還原形成SEI。隨后,陰離子主導(dǎo)分解形成的SEI會(huì)削弱電極對(duì)Li+-DME-DFOB-或Li+-DME-FSI-配合物的供電子能力,提高了電解液的穩(wěn)定性。基于上述對(duì)電解液穩(wěn)定性的分析,成功地闡明了微米Sb負(fù)極的優(yōu)異性能的原因。
相比之下,在3.75 M LiFSI in DME (Li+[DME]2.56[FSI-])電解液中,由于沒有LiDFOB分散DME溶劑參與解離溶劑化,參與解離溶劑化LiFSI的DME溶劑分子數(shù)量增多,第一溶劑化層重疊的程度較有添加劑的減少。換言之,Li+-FSI-的相對(duì)距離(d2)略長于Li+[DME]2.26[FSI-]0.88[DFOB-]0.12中Li+-FSI-的相對(duì)距離(d1,d1 < d2)。
同樣的,在圖6b'的界面模型中Li+與FSI-之間的相對(duì)距離(d2')也相對(duì)遠(yuǎn)一些,即d2' > d1'。在去溶劑化過程中,F(xiàn)SI-雖然也會(huì)優(yōu)先接收電極上的電子參與形成SEI,但與DFOB-衍生的SEI膜相比,F(xiàn)SI-誘導(dǎo)生成的SEI膜并不穩(wěn)定,不足以有效的削弱電極的供電子能力。
這樣會(huì)導(dǎo)致Li+-DME或Li+-2DME-FSI-配合物可以繼續(xù)得到電子發(fā)生分解,造成SEI中C含量的增加,在電極表面形成富C和F有機(jī)物的SEI。以上因素共同影響,使得微米Sb負(fù)極在幾次循環(huán)后就不能正常工作。在Li+[DME]9.6[FSI-]0.88[DFOB-]0.12電解液中進(jìn)一步證實(shí)了LiDFOB添加劑的作用。當(dāng)LiFSI濃度降低到1.0 M時(shí),由于參與溶劑化的DME溶劑分子數(shù)量足夠多,Li+溶劑化結(jié)構(gòu)(d3,f3)和界面模型(d3',f3')中Li+和FSI-、DFOB-的相對(duì)距離最長(圖6c,c')。
此外,Li+-2DME-FSI-配合物的ΔE值為0.0745 Hartte,小于Li+-DME-FSI-配合物的0.0770 Hartte(圖6f),表明Li+[DME]9.6[FSI-]0.88[DFOB-]0.12電解液的穩(wěn)定性相比高濃度電解液要差。在去溶劑化過程中,DFOB-和DME溶劑分子會(huì)很容易在電極表面得到從電極上傳遞的電子發(fā)生分解,在電極表面生成富B、C和F物質(zhì)的SEI。
生成的SEI可以在一定程度上抑制電極的供電子能力,相對(duì)減少DME的分解,使得微米Sb負(fù)極可以維持基本的循環(huán)。然而,在Li+[DME]9.6[FSI-]0.88[DFOB-]0.12電解液中,由于DME溶劑能充分解離、溶劑化鋰鹽,DME溶劑分子被Li+的極化程度較高,會(huì)不能有效地抑制循環(huán)過程中電解液的分解,在電極表面生成富含大量有機(jī)物的SEI,導(dǎo)致電極阻抗逐漸變大。
以上結(jié)果進(jìn)一步表明提高鋰鹽濃度和引入LiDFOB添加劑,可以提高電解液穩(wěn)定性,改善電極性能。此外,選擇合適的陰離子對(duì)維持電解液的穩(wěn)定性也很重要。如圖6d,d'所示,在Li+[DME]2.26[TFSI-]0.88[DFOB-]0.12電解液中,Li+溶劑化結(jié)構(gòu)和界面模型中Li+-TFSI-(d4,d4')的相對(duì)距離與Li+[DME]2.26[FSI-]0.88[DFOB-]0.12電解液中的相對(duì)距離要遠(yuǎn)一些,而Li+-DFOB-(f4,f4')的相對(duì)距離則相當(dāng)。
在去溶劑化過程中,由于Li+-DME-DFOB-配合物較低的ΔE值,使得DFOB-更容易在電極表面發(fā)生分解,并形成富B和F物質(zhì)的SEI。雖然生成的SEI能夠削弱電極的供電子能力,但由于Li+-DME-TFSI-配合物的ΔE值(0.0760 Hartte)低于Li+-DME-FSI-配合物的,且Li+對(duì)DME溶劑分子的極化程度相對(duì)較大,造成電解液的分解,最終導(dǎo)致電池容量的衰減。
以上研究結(jié)果表明,Li+溶劑化結(jié)構(gòu)及其界面模型與SEI能有效地改善微米Sb負(fù)極電化學(xué)性能。其中SEI可以有效地降低電極的給電子能力,減少電解液的分解,進(jìn)而提高電解液的穩(wěn)定性。
7.鋰離子全電池應(yīng)用
為了進(jìn)一步驗(yàn)證所設(shè)計(jì)電解液的優(yōu)勢(shì),組裝了以微米Sb為負(fù)極,LiFePO4做正極,3.75 M LiFSI/0.5 M LiDFOB in DME做電解液液的鋰離子全電池,如圖7所示。該電池在0.5C(1C = 150 mA/g)電流密度下表現(xiàn)出了良好的循環(huán)性能,經(jīng)100次循環(huán),容量保持在78.4 mAh/g,容量保持率為54%。
此外,在0.1、0.2、0.5、1、2和5C電流密度下,容量分別為155、151、141和126 mAh/g,表明了全電池具有優(yōu)異的倍率性能。上述結(jié)果充分證明了微米Sb負(fù)極在3.75 M LiFSI/0.5 M LiDFOB in DME電解液中的高穩(wěn)定性和可逆性。
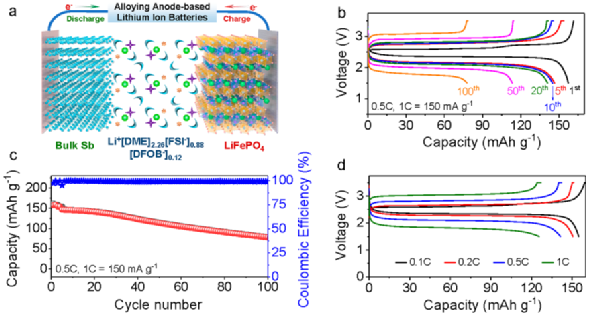
圖7. 全電池性能。
【結(jié)論】
該工作以微米Sb負(fù)極為研究對(duì)象,設(shè)計(jì)了一款新的基于DME的電解液,以認(rèn)識(shí)溶劑化結(jié)構(gòu)衍生的界面模型和SEI對(duì)電極性能的決定性影響。研究發(fā)現(xiàn),鋰鹽濃度、種類和添加劑對(duì)于特定SEI和界面模型的形成都至關(guān)重要,最終影響電極的循環(huán)穩(wěn)定性。尤其,該工作從溶劑化結(jié)構(gòu)、界面模型到最終形成的SEI的動(dòng)態(tài)演化過程全面闡述了電解液組分尤其添加劑的作用。該研究為通過調(diào)節(jié)溶劑化結(jié)構(gòu)和SEI的協(xié)同方法設(shè)計(jì)電解液提供了指導(dǎo)。
審核編輯:劉清
-
電解液
+關(guān)注
關(guān)注
10文章
849瀏覽量
23155 -
固體電解質(zhì)
+關(guān)注
關(guān)注
0文章
46瀏覽量
8406
原文標(biāo)題:應(yīng)化所明軍研究員電解液融合篇:溶劑化結(jié)構(gòu)與SEI膜的是是非非
文章出處:【微信號(hào):清新電源,微信公眾號(hào):清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請(qǐng)注明出處。
發(fā)布評(píng)論請(qǐng)先 登錄
相關(guān)推薦
調(diào)控磷酸酯基阻燃電解液離子-偶極相互作用實(shí)現(xiàn)鈉離子軟包電池安全穩(wěn)定運(yùn)行
貼片鋁電解電容的封裝材質(zhì)型號(hào)有哪些?

水系電解液寬電壓窗口設(shè)計(jì)助力超長壽命水系鈉離子電池
ADS1208一款二階AE調(diào)制器數(shù)據(jù)表

鎳氫電池的電解液是什么
高壓電解電容虛標(biāo)原因,高壓電解電容虛標(biāo)怎么判斷
新宙邦擬在美國投建10萬噸/年電解液項(xiàng)目
瑞能半導(dǎo)體推出一種帶隙大于傳統(tǒng)硅基肖特基二極管的半導(dǎo)體器件

液位傳感器監(jiān)測(cè)鉛酸電池電解液液位

非質(zhì)子型弱配位電解液實(shí)現(xiàn)無腐蝕超薄鋅金屬電池






 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論