") SC22 | 解析基因組的“語言”:戈登貝爾獎決賽選手使用大型語言模型來預測新冠病毒變異株
SC22 | 解析基因組的“語言”:戈登貝爾獎決賽選手使用大型語言模型來預測新冠病毒變異株
來自美國阿貢國家實驗室、NVIDIA、芝加哥大學等組織機構(gòu)的研究員開發(fā)了一個處理基因組規(guī)模數(shù)據(jù)的先進模型,并入圍戈登貝爾 COVID-19 研究特別獎決賽
這一戈登貝爾特別獎旨在表彰基于高性能計算的 COVID-19 研究。一位決賽入圍選手教會了大型語言模型(LLMs)一種新的語言——基因序列,使這些模型能夠提供基因組學、流行病學和蛋白質(zhì)工程方面的洞察。
這項開創(chuàng)性的成果發(fā)表于 10 月,是由來自美國阿貢國家實驗室、NVIDIA、芝加哥大學等組織機構(gòu)的二十多名學術(shù)和商業(yè)研究員合作完成。
該研究團隊訓練了一個 LLM 來追蹤基因突變,并預測需要關(guān)注的 SARS-CoV-2(導致 COVID-19 的病毒)變異株。雖然迄今為止大多數(shù)應(yīng)用于生物學的 LLM 都是在小分子或蛋白質(zhì)的數(shù)據(jù)集上訓練的,但這一項目是在原始核苷酸序列(DNA 和 RNA 的最小單位)上訓練的首批模型之一。
負責帶領(lǐng)該項目的阿貢國家實驗室計算生物學家 Arvind Ramanathan 表示:“我們假設(shè)從蛋白質(zhì)水平到基因水平的數(shù)據(jù)有助于我們構(gòu)建出更易于理解新冠病毒變異株的模型。通過訓練模型去追蹤整個基因組及其進化過程中的所有變化,我們不僅能夠更好地預測 COVID,還能預測已掌握足夠基因組數(shù)據(jù)的任何疾病。”
戈登貝爾獎被譽為 HPC 領(lǐng)域的諾貝爾獎。今年的戈登貝爾獎將在本周的 SC22 上由美國計算機協(xié)會頒發(fā)。該協(xié)會代表著全球約 10 萬名計算領(lǐng)域的專家,自2020年開始向使用 HPC 推進 COVID-19 研究的杰出研究員頒發(fā)特別獎。
在一種只有四個字母的語言上
訓練大型語言模型
長期以來,LLM 一直在接受人類語言的訓練,這些語言通常由幾十個字母組成,可以排列組合成數(shù)萬個單詞,并連接成長句和段落。而生物學語言只有四個代表核苷酸的字母,即 DNA 中的 A、T、G 和 C,或 RNA 中的 A、U、G 和 C。這些字母按不同順序排列成基因。
雖然較少的字母看似會降低 AI 學習的難度,但實際上生物學語言模型要復雜得多。這是因為人類的基因組由超過 30 億個核苷酸組成,而冠狀病毒的基因組由大約 3 萬個核苷酸組成,因此很難將基因組分解成不同、有意義的單位。
Ramanathan 表示:“在理解基因組這一‘生命代碼’的過程中,我們所面對的一個主要挑戰(zhàn)是基因組中的龐大測序信息。核苷酸序列的意義可能會受另一序列的影響,以人類的文本做類比,這種影響的范圍不僅僅是文本中的下一句話或下一段話,而是相當于一本書中的整個章節(jié)。”
參與該項目協(xié)作的 NVIDIA 研究員設(shè)計了一種分層擴散方法,使 LLM 能夠?qū)⒓s 1500 個核苷酸的長字符串當作句子來處理。
論文共同作者、NVIDIA AI 研究高級總監(jiān)、加州理工學院計算+數(shù)學科學系布倫講席教授 Anima Anandkumar 表示:“標準語言模型難以生成連貫的長序列,也難以學習不同變異株的基本分布。我們開發(fā)了一個在更高細節(jié)水平上運作的擴散模型,該模型使我們能夠生成現(xiàn)實中的變異株,并采集到更完善的統(tǒng)計數(shù)據(jù)。”
預測需要關(guān)注的新冠病毒變異株
該團隊首先使用細菌和病毒生物信息學資源中心的開源數(shù)據(jù),對來自原核生物(像細菌一樣的單細胞生物)超過 1.1 億個基因序列進行了 LLM 預訓練,然后使用 150 萬個高質(zhì)量的新冠病毒基因組序列,對該模型進行微調(diào)。
研究員還通過在更廣泛的數(shù)據(jù)集上進行預訓練,確保其模型能夠在未來的項目中推廣到其他預測任務(wù),使其成為首批具備此能力的全基因組規(guī)模的模型之一。
在對 COVID 數(shù)據(jù)進行了微調(diào)后,LLM 就能夠區(qū)分病毒變異株的基因組序列。它還能夠生成自己的核苷酸序列,預測 COVID 基因組的潛在突變,這可以幫助科學家預測未來需要關(guān)注的變異株。
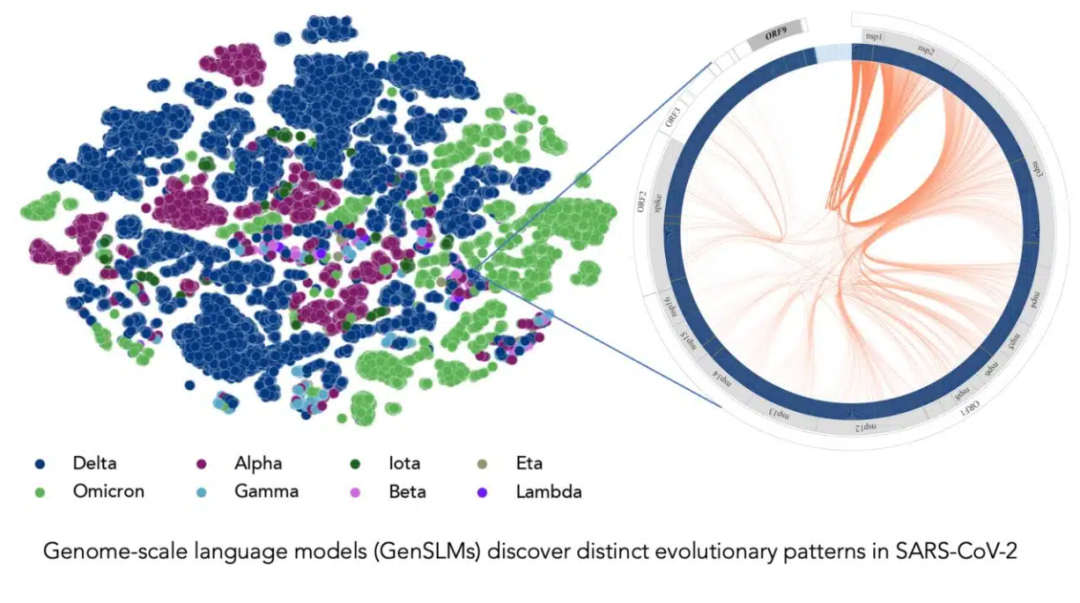
在長達一年時間內(nèi)積累的 SARS-CoV-2 基因組數(shù)據(jù)的訓練下,該模型可以推斷出各種病毒株之間的區(qū)別。左邊的每個點對應(yīng)一個已測序的 SARS-CoV-2 病毒株,并按變異株顏色編碼。右圖放大了該病毒的一個特定毒株,它捕捉到了該毒株特有的病毒蛋白進化耦合關(guān)系。圖片由美國阿貢國家實驗室的 Bharat Kale、Max Zvyagin 和 Michael E. Papka 提供。
Ramanathan 表示:“大多數(shù)研究員一直在追蹤新冠病毒突刺蛋白的突變,尤其是與人類細胞結(jié)合的域。但病毒基因組中還有其他蛋白質(zhì)也會經(jīng)歷頻繁的突變,所以了解這些蛋白質(zhì)十分重要。”
論文中提到,該模型還可以與 AlphaFold、OpenFold 等常見的蛋白質(zhì)結(jié)構(gòu)預測模型整合,幫助研究員模擬病毒結(jié)構(gòu),研究基因突變?nèi)绾斡绊懖《靖腥酒渌拗鞯哪芰ΑpenFold 是 NVIDIA BioNeMo LLM 服務(wù)中包含的預訓練語言模型之一。NVIDIA BioNeMo LLM 服務(wù)面向的是致力于將 LLM 應(yīng)用于數(shù)字生物學和化學應(yīng)用的開發(fā)者。
利用 GPU 加速超級計算機
大幅加快 AI 訓練速度
該團隊在由 NVIDIA A100 Tensor Core GPU 驅(qū)動的超級計算機上開發(fā) AI 模型,包括阿貢國家實驗室的 Polaris、美國能源部的 Perlmutter 以及 NVIDIA 的 Selene 系統(tǒng)。通過擴展到這些強大的系統(tǒng),他們在訓練中實現(xiàn)了超過 1500 exaflops 的性能,創(chuàng)建了迄今為止最大的生物語言模型。
Ramanathan 表示:“我們?nèi)缃裉幚淼哪P陀卸噙_ 250 億個參數(shù),預計這一數(shù)量未來還會大幅增加。模型的尺寸、基因序列的長度、以及所需的訓練數(shù)據(jù)量,都意味著我們的確需要搭載數(shù)千顆 GPU 的超級計算機來完成復雜的計算。”
研究員估計,訓練一個具有 25 億參數(shù)的模型版本,需要約 4000 個 GPU 耗時一個多月。該團隊已經(jīng)在研究用于生物學的 LLM,在公布論文和代碼之前,他們在這個項目上已耗時約四個月。GitHub 頁面上有供其他研究員在 Polaris 和 Perlmutter 上運行該模型的說明。
NVIDIA BioNeMo 框架可在 NVIDIA NGC 中心上的 GPU 優(yōu)化軟件中搶先體驗。該框架將幫助研究員在多個 GPU 上擴展大型生物分子語言模型。作為 NVIDIA Clara Discovery 藥物研發(fā)工具集的一部分,該框架將支持化學、蛋白質(zhì)、DNA 和 RNA 數(shù)據(jù)格式。
即刻點擊“閱讀原文”或掃描下方海報二維碼,收下這份 GTC22 精選演講合集清單,在NVIDIA on-Demand 上點播觀看主題演講精選、中國精選、元宇宙應(yīng)用領(lǐng)域與全球各行業(yè)及領(lǐng)域的最新成果!
原文標題:SC22 | 解析基因組的“語言”:戈登貝爾獎決賽選手使用大型語言模型來預測新冠病毒變異株
文章出處:【微信公眾號:NVIDIA英偉達企業(yè)解決方案】歡迎添加關(guān)注!文章轉(zhuǎn)載請注明出處。
-
英偉達
+關(guān)注
關(guān)注
22文章
3780瀏覽量
91217
原文標題:SC22 | 解析基因組的“語言”:戈登貝爾獎決賽選手使用大型語言模型來預測新冠病毒變異株
文章出處:【微信號:NVIDIA-Enterprise,微信公眾號:NVIDIA英偉達企業(yè)解決方案】歡迎添加關(guān)注!文章轉(zhuǎn)載請注明出處。
發(fā)布評論請先 登錄
相關(guān)推薦
大語言模型開發(fā)框架是什么
大語言模型開發(fā)語言是什么
如何利用大型語言模型驅(qū)動的搜索為公司創(chuàng)造價值

【《大語言模型應(yīng)用指南》閱讀體驗】+ 基礎(chǔ)知識學習
基于CPU的大型語言模型推理實驗

基于神經(jīng)網(wǎng)絡(luò)的語言模型有哪些
大語言模型(LLM)快速理解

了解大型語言模型 (LLM) 領(lǐng)域中的25個關(guān)鍵術(shù)語





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評論