 如何在云計算平臺上完成分子對接
如何在云計算平臺上完成分子對接
分子對接的基本原理就是把配體分子放在受體活性位點的位置,然后按照幾何互補和能量互補的原則來實時評價配體與受體相互作用的優劣,最終找到這兩個分子之間最佳的結合模式。
分子對接分為剛性、半柔性和柔性對接。不同的對接軟件又可以分為:商業軟件和學術軟件,而分子對接的計算結果,則體現為打分函數的不同。目前用的比較多的分子對接軟件有:AutoDock、AutoDock Vina、LeDock、rDock,這些都是學術軟件;商業軟件有:Glide、GOLD、MOE Dock、Surflex-Dock、LigandFit、FlexX等等。
做分子對接,一般對接的分子量都很大,到底有多大?就是很大!
我來舉個例子,通常分子對接的采樣,都是百萬到千萬級別的分子
而事實上可用于藥物發現的有機分子有多少?
超過10的60次方!
我們取的只是滄海一粟罷了。
這么大的對接量,對算力的需求肯定少不了呀!
那怎么辦呢?
具體,我以AutoDock-Vina分子對接為例。
AutoDock-Vina 是用于分子對接和虛擬篩選的開源程序,由Scripps研究所分子圖形實驗室的Oleg Trott博士設計和實現,是目前使用最為廣泛的分子對接軟件之一。此外,Vina可以利用系統上的多個CPU或CPU內核來顯著縮短運行時間;北鯤云超算平臺提供了豐富的cpu,gpu資源,此次教程以STAT3靶點的晶體結構與其原配體為例進行分子對接的詳細步驟說明,就在北鯤云上演示。
準備工作
蛋白晶體結構由PDB數據庫下載,PDB編號為6NJS。首先需要通過其他軟件如Pymol、Glide、DS等去除蛋白中的水分子,刪除多余的鏈,并把原配體分子提離出來。單獨的蛋白文件和配體文件均保存為.pdb格式。
設置工作目錄:
為了計算的方便以及后續文件的方便查找,我們首先設置一個工作目錄,要注意文件路徑需全為英文,否則會導致程序出現錯誤。
我這里在C盤中設立文件夾AutoDock,以后的計算均可保存在該文件夾中。
為了將不同的計算結果分開,我們再創建一個文件夾6NJS在AutoDock文件夾下。
AutoDock vina的計算需要用到兩個程序,即vina.exe和vina_split.exe。其中vina.exe是用來做對接,vina_split.exe用來分割對接構象結果。
在運算之前,從下載好的文件夾中復制這兩個程序至6NJS文件夾中。同時將保存的蛋白及原配體的.PDB文件也放置在該文件夾中,其中6njs.pdb為蛋白文件,KQV701.pdb為原配體文件。
受體和配體的預處理
打開預裝好AutoDockTools的windows工作站,在菜單欄點擊File>ReadMolecule打開6NJS文件夾中的6njs.pdb。
-受體準備-
①除水:在準備工作中蛋白中水分子已被刪除,這一步即可省略。
②加氫:Edit>Hydrogens>Add>OK
③計算電荷:Edit>Charges>ComputeGasteiger
④添加原子類型:Edit->Atoms->AssignAD4 type
⑤保存為.pdbqt文件:File->Save->WritePDBQT,此時可在6NJS文件夾下看到多了一個“6njs.pdbqt”文件
-配體準備-
點擊鼠標右鍵>delete 在Dashboard中把受體分子刪除。通過ADT菜單欄Ligand->Input->Open打開KQV701.pdb文件,彈出一個對話框,點擊確定
①調整電荷:彈出窗口提示配體分子的總電荷數不是整數。點擊Edit->Charges->Check Totals on Residues>Spread ChargeDeficit over all atoms in residue>Dismiss
②判定配體的root:ADT菜單欄Ligand>Torsion Tree>DetectRoot
③選擇配體可扭轉的鍵:Ligand>TorsionTree>Choose Torsions>Done,表示該分子32個鍵中有13個可旋轉的。
其中紅色表示不可旋轉的鍵,綠色表示可旋轉鍵,紫色表示不可扭轉,通常為肽鍵。如果要設置某個鍵不可扭轉,那么先按住shift鍵,然后鼠標單擊即可(顏色變成紫色)。
④保存為.pdbqt文件:ADT菜單欄Ligand->Output->Saveas PDBQT,此時可在6NJS文件夾下看到多了一個“KQV701.pdbqt”文件
創建對接信息文件
在6NJS文件夾中創建一個配置文件:6njs.conf,這個文件里面寫上用于對接的詳細參數:
receptor = 6njs.pdbqt
ligand = KQV701.pdbqt
center_x = 13.24
center_y = 54.43
center_z = 0.27
size_x = 20.6
size_y = 31.1
size_z = 23.1
energy_range = 4
exhaustiveness = 12
num_modes = 10
receptor:表示受體分子的路徑
ligand:表示配體分子的路徑
center_x,center_y,center_z:表示活性位點盒子中心的坐標,我們這里以配體擴張法定義對接盒子,即以原配體所在位置為中心向外擴張一定的范圍。如果沒有原配體可以通過文獻查找或工具預測的方法獲得。
size_x,size_y,size_z:指定對接盒子的大小。這里設置的大小至少要包裹活性位點的空腔,但不宜設置過大,負責對接結果不準確,具體的設置可根據對接結果的好壞重新調整。
energy_range:與最優結合模型相差的最大能量值,單位是kcal/mol。我們設置為4則表示vina最多計算到與最優模型的能量差值為4就終止計算了。
exhaustiveness:用來控制對接的細致程度,默認值是8,設置值與電腦的配置相關,影響計算時間。
num_modes:最多生成多少個模型。此時,6NJS文件夾中應該包括配體分子,受體分子,參數文件,vina程序共7個文件。
執行計算
打開北鯤云超算平臺,進入控制臺
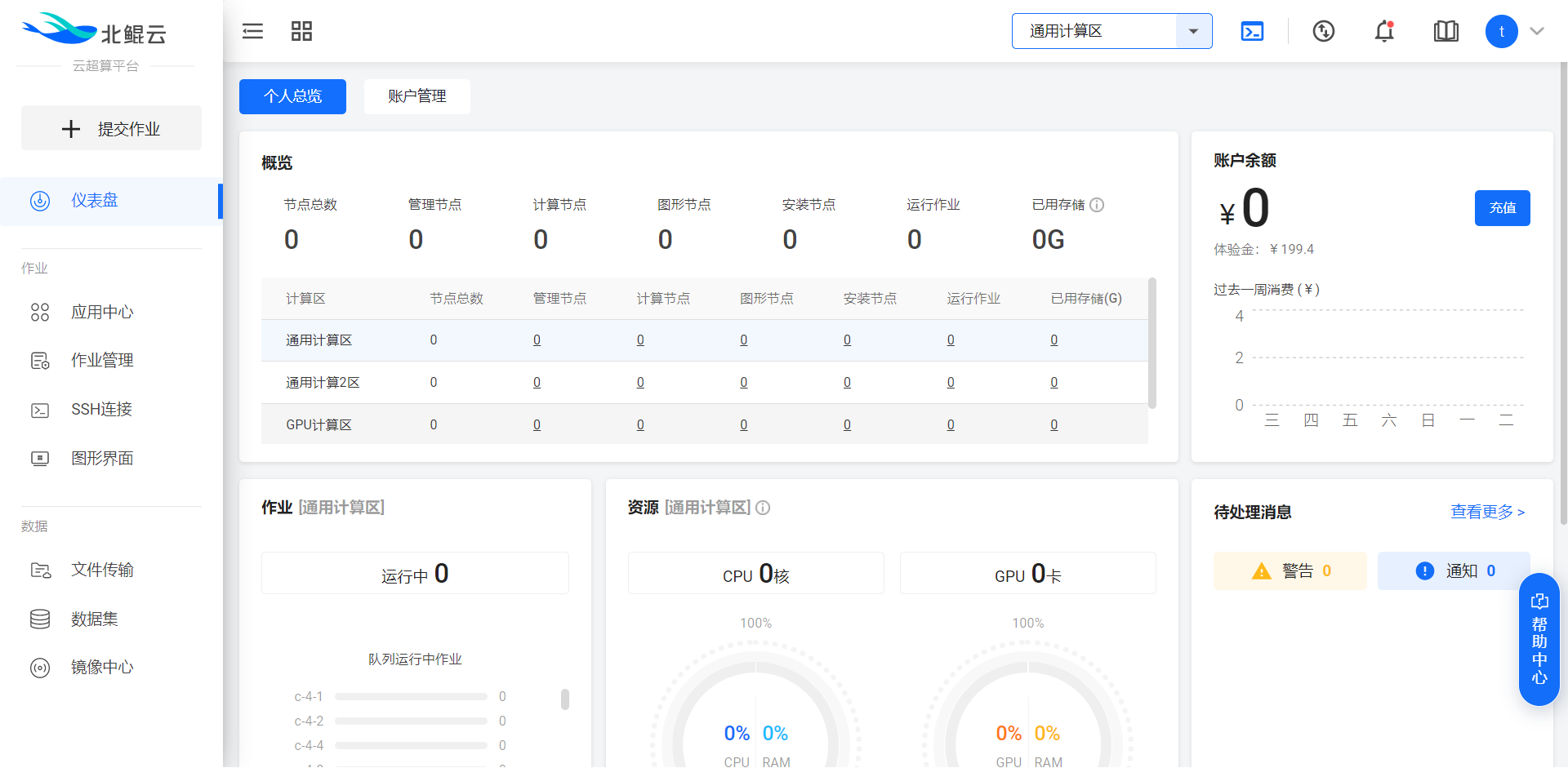
這里選擇SSH連接,通過SSH鏈接啟動一個管理節點,并連接進入管理節點。
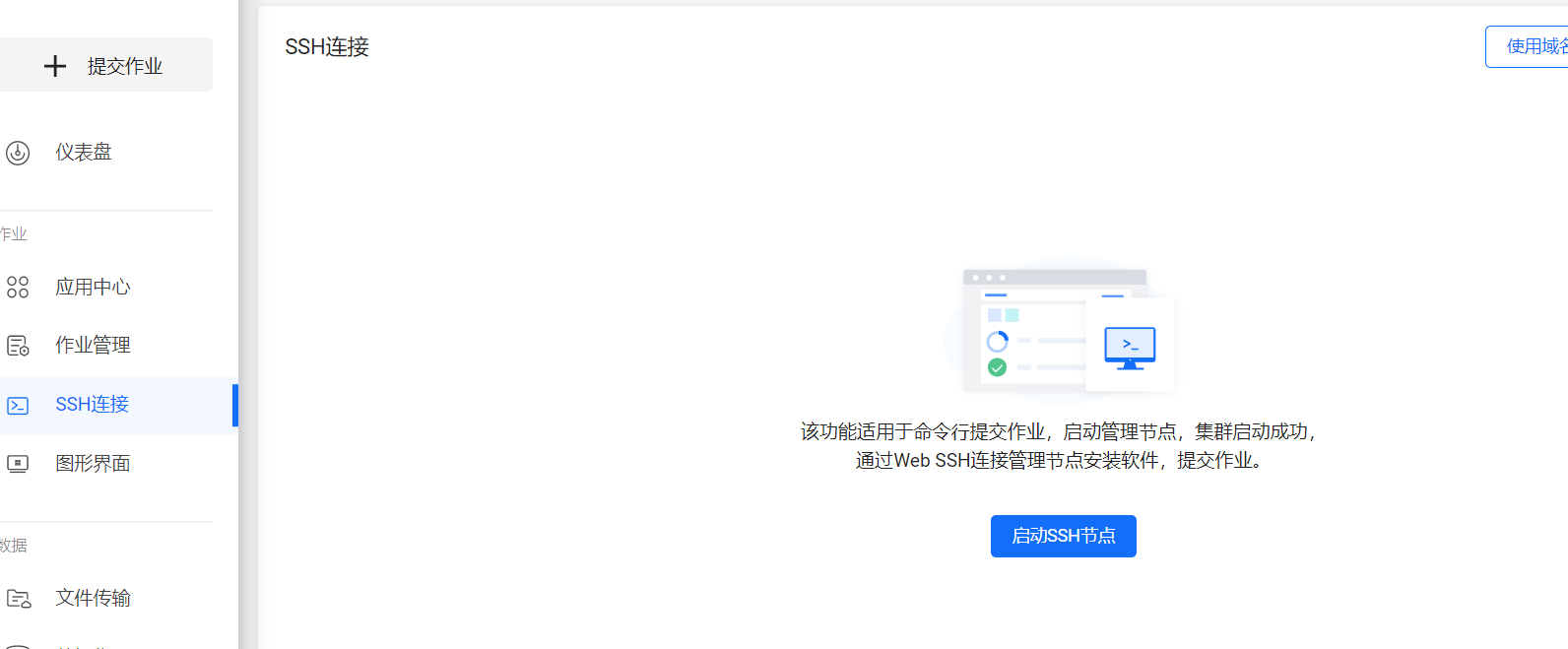
通過win+R進入運行窗口,輸入“cmd”進入命令行窗口,此時默認文件夾一般為C盤,
再輸入
CDC:AutoDock6NJS
回車,進入該文件夾。
輸入
vina--config6njs.conf
回車,即可執行分子對接計算。
等待計算完成,一共得到10個模型結果,包括對接結合能分數,RMSD值,我們可以看到有多個結果的RMSD都小于2埃,說明本次分子對接結果還是比較可靠的。
在北鯤云超算平臺完成分子對接計算的過程還是比較簡單的,只要按照上述步驟即可完成,中途如果遇到問題可以隨機聯系我們的技術支持,技術支持隨時在線解決大家的疑惑。
除了簡便的操作之外,在平臺上還有海量資源供大家選擇,不用擔心要排隊或者ddl趕不上啦!
審核編輯黃昊宇
-
云計算
+關注
關注
39文章
7848瀏覽量
137636
發布評論請先 登錄
相關推薦
深度評測:云計算平臺的優勢和不足
云計算平臺的最佳實踐
云計算平臺層(PaaS)指的是什么?常見的應用場景盤點
通過機智云平臺電腦網頁控制設備的指南

如何在AT COMMAND的方式下快速的完成Lierda NB861/MB961/MB261模組與OneNET平臺的對接工作?
通過MQTT網關快速對接工業物聯網云平臺
EM儲能網關 ZWS智慧儲能云應用(2) — 建模介紹

能在Meteor Lake平臺上使用SDK 3.5嗎?
通過MQTT網關快速對接工業物聯網云平臺

淺談云計算平臺的電動汽車充電樁設計與實現





 工商網監
工商網監
評論