 阿貢實驗室Chem發文再談Zn2+配位環境
阿貢實驗室Chem發文再談Zn2+配位環境
研究背景
隨著儲能技術逐漸成為現代社會不可或缺的一部分,不斷增長的儲能需求對各種電池材料提出了重要的科學挑戰。尋找能夠超越當前鋰離子電池的新型電化學體系,包括水系、非水系和混合溶劑系統,已成為當前研究的熱點問題。但目前已經越來越清楚,無論所研究的具體化學性質如何,理解并調控陽離子在界面的溶劑化結構對于提高電解液穩定性和電極可逆性至關重要。這種控制可以通過調節溶劑和陰離子的濃度和化學成分來實現。然而,對于混合電解液系統中復雜相互作用的清晰理解以及如何調整這種相互作用以產生特定的溶劑化結構,是一個急需解決的問題。
因此,阿貢實驗室的科學家在文章中指出,多價離子(如Ca2+、Mg2+和Zn2+)比Li+具有更大的電荷密度,因此在電解液中有強烈的離子配對和聚集的驅動力。在低介電常數溶劑中,這些離子配對的相互作用尤其難以打破,因此線性和環狀醚這樣還原穩定性高的溶劑,通常被用于多價系統中。迄今為止,許多多價電解液都依賴于具有顯著氯離子(Cl?)濃度的混合陰離子電解液,這些電解液通常用于驅動可逆氧化還原過程的陽極。但是,這些系統與氧化物陰極不兼容,因為在高電壓下會發生寄生反應。
最近的研究表明,Cl?在這些系統中的主要作用是降低金屬溶解的過電位,這是以更高的金屬沉積過電位為代價的。引入弱配位陰離子,如雙(三氟甲烷磺酰)亞胺(TFSI?),可以產生協同效應,從而實現可逆的金屬沉積和溶解。這種協同效應是由于鹵離子-陽離子絡合物與TFSI?-陽離子絡合物的結合強度之間的差異所引起的,其中弱配位的TFSI?-陽離子絡合物有助于陽離子脫溶,從而可以實現低過電位的金屬沉積,而強配位的Cl?-金屬絡合物有助于陽離子溶解,從而可以實現低過電位的金屬溶解。這些相互作用在幾種工作陽離子(例如Cu2+、Zn2+和Mg2+)中都是通用的,表明陰離子結合強度可以作為系統性能的描述符。
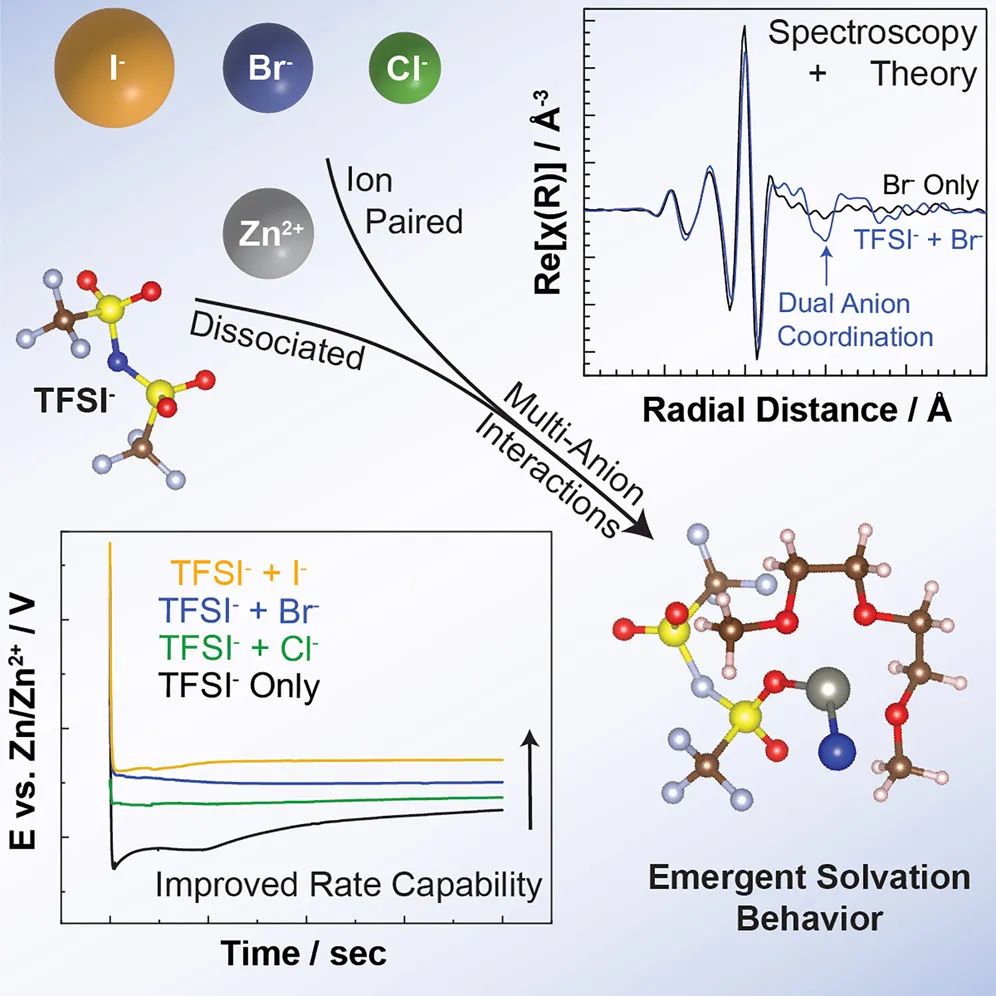
圖形摘要
成果簡介
在這篇研究論文中,作者將實驗光譜學與密度泛函理論相結合,來表征當鋅離子被兩種不同的陰離子溶劑化時出現的溶劑化行為。鋅和多種陰離子之間的接觸離子對的形成表明能夠在電極表面上實現新的還原和氧化行為。這些結果為新型電解液設計提供了藍圖,在這種設計中,可以根據需要來設計電化學性能。
文章亮點
(1)鹵化物和有機陰離子的混合誘導出Zn2+配位環境;
(2)電解液體相中配位直接影響在電極上的沉積動力學;
(3)鹵化物締合強度的降低與氧化還原活性的增加有關。
圖文導讀
本體溶劑化表征
通過XANES和EXAFS測量,描述了Zn2+溶液絡合物的局部結構。在對Zn2+陽離子周圍的局部結構進行初步分析時,使用了溶解在二甘醇二甲醚中的Zn2+基電解液,該電解液含有純ZnX2、純ZnTFSI2和兩者1:1混合(圖1A)。在鹵化物中,作者在XANES和EXAFS中發現了一致的結果,描述了在TFSI?和鹵化物陰離子存在的情況下Zn–鹵素離子配對和獨特的配位效應。在每個系列中,ZnTFSI2電解液(黑線,圖1A–1C)顯示出一條與先前關于Zn2+八面體絡合物的文獻一致的單一、寬的白線。對ZnTFSI2電解液的EXAFS區域的分析表明,Zn–二甘醇第一殼層配位占主導地位。表明當溶解在線性醚溶劑中時,TFSI?在本體第一殼層Zn2+配位中只起到很小的作用(如果有的話)。ZnTFSI2電解液EXAFS與僅源自二甘醇配位分子的模型光電子散射路徑的擬合在數據建模中是有效的,而包含來自TFSI?配位的Zn–S和Zn–O–S相關性并沒有從統計學上改善擬合。
采用拉曼光譜測量對 TFSI-振動模式的擾動,這些振動模式對陰離子配位和離子配對敏感。僅含 ZnTFSI 2 的電解液的測量在 740 cm-1處產生強峰。(圖 1D),與之前關于鋰基、鋅基和鎂基電解液的報道一致。可以使用 Voigt 函數將該峰包絡解卷積為三個獨立的貢獻,并使用先前報告的值固定擬合參數。主要的“游離 TFSI”峰出現在大約 740 cm -1處,其中“游離”意味著 TFSI -陰離子不與任何陽離子強相關。在 ~742 cm -1處的第二個中頻峰以前被歸因于單齒接觸離子對 (CIP) TFSI -配位。~750 cm ?1峰對應的第三環境也是一種 CIP 溶劑化結構,以前被歸因于雙齒 TFSI -配位。總體而言,拉曼分析強烈表明存在大量混合陰離子 CIP。不同電解液溶液(ZnX 2、ZnTFSI 2和 ZnX 2 + ZnTFSI 2 )之間傅立葉變換 EXAFS(圖 1 E)的比較(X = Br, I)——明確顯示了雙陰離子電解液系統中存在的額外信號,這些信號不能完全描述為 Zn-X 或 Zn-二甘醇二甲醚相關性,進一步支持混合陰離子對的形成。
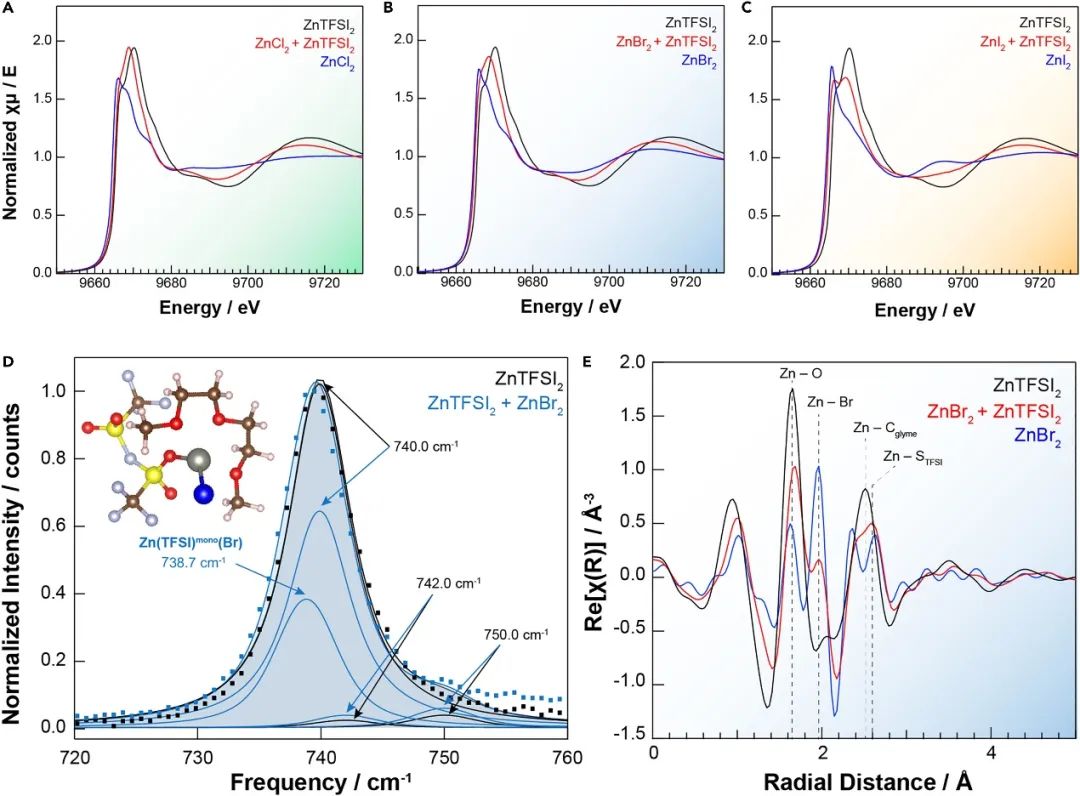
圖1 混合陰離子電解液的X 射線和振動光譜表征。
雙陰離子 ZnX 2 + ZnTFSI 2的 EXAFS 建模揭示了額外的、意想不到的散射成分,這些成分是由 Zn-TFSI 相關性引起的,這些成分被發現在第一個溶劑化結構中具有統計相關性。ZnX 2 + ZnTFSI 2電解液的 Zn K-edge 和 Br 或 I K-edge 同時擬合顯示 Zn 邊緣超過 2.5 ? 的顯著信號需要在大約 3.20 ? 處包含 Zn-S 散射路徑(圖2 A)。在探測 Br K 邊(圖 2 C)和 I K 邊(圖 2 B)時,專門針對雙陰離子電解液發現了額外的相關性。在Br的K 邊的2.75 和 3.5 ? 之間的徑向距離處發現的這些特征表明 TFSI -配位改變了 Br -周圍的局部結構,并表明 Br-TFSI 散射路徑可能源自單個 Zn 2+協調結構。
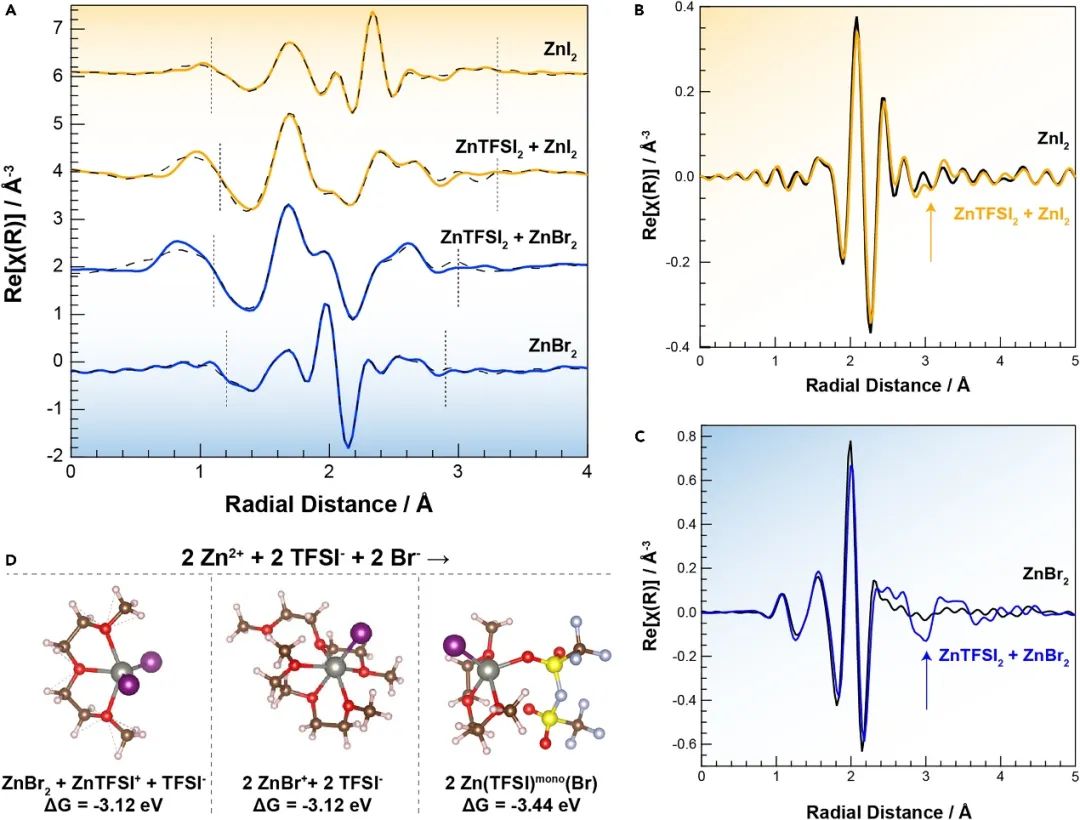
圖2. 絡合自由能的EXAFS擬合和DFT分析。
為確定此類結構是否在熱力學上有利,利用DFT計算手段,并考慮了多種途徑。計算表明支持實驗觀察到的溶劑化行為的幾種路徑,總結在圖 2 D 中。具體來說,離子配對 Zn(TFSI) (Br) 絡合物的形成在所有考慮的結果中被發現在熱力學上是最有利的 (ΔG = - 3.44 eV),與 EXAFS 和拉曼在混合鹽電解液中觀察到的 Zn-TFSI 離子對一致。Zn–Br 和 Zn–TFSI 離子對混合物的形成(即 2 Zn 2++ 2 TFSI?+ 2 Br? → ZnBr 2 + ZnTFSI ++ TFSI ?)也被發現在反應能方面具有競爭力( ΔG = ?3.12 eV)。
2.陰離子協同效應引起的電化學反應趨勢
測量了混合 TFSI-Br 和 TFSI-I 電解液的循環伏安圖,并與之前在 TFSI-Cl 系統中的結果進行了比較(圖 3A )。我們注意到,在所有情況下,都選擇玻碳作為工作電極,以避免在 Zn 金屬成核和生長之前鹵化物的特定吸附等過程的混雜效應。與 TFSI-Cl 系統類似,TFSI-Br 和 TFSI-I 電解液均表現出多個還原和氧化峰,與多個 Zn 2+的參與一致沉積/溶解半反應中的配位結構。具體來說,在所有電解液的正向掃描中觀察到兩個還原峰,這與低過電位(約 1.95 V)下富含 TFSI 的配位絡合物和較高過電位(1.80 V)下富含鹵化物的配位絡合物的沉積一致。有趣的是,對于含 Br 和 I 的電解液,在反向掃描中有額外的還原峰,觀察到 I -相對于 Br -的額外還原電流。這些特征表明,與含 Cl 的電解液相比,Zn 金屬沉積可能存在更多途徑,可能來自更復雜和多樣化的溶液形態。
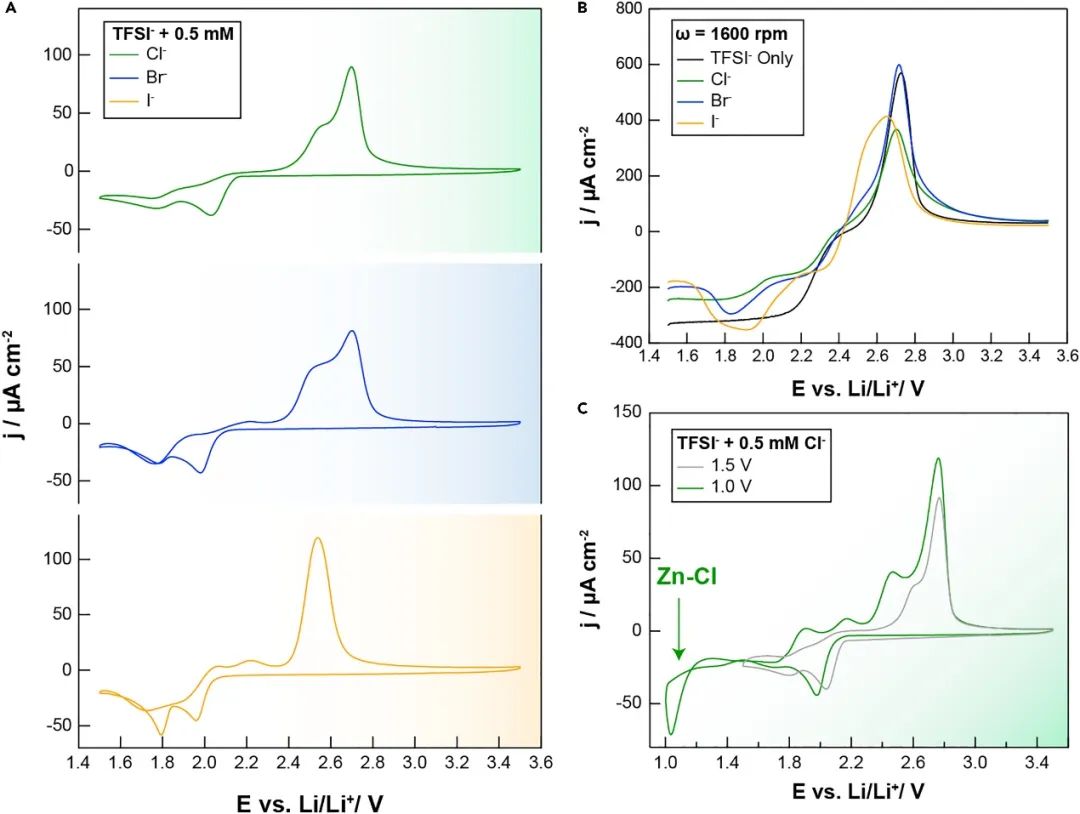
圖3. 混合陰離子電解液活性的電化學分析。
TFSI-Cl 和 TFSI-Br 電解液也觀察到兩個氧化峰,在 2.50 V處具有低過電勢特征。與 Zn 溶解到富含鹵化物的配位環境和處的高過電位溶解峰一致。兩個峰的相對大小也隨著鹵化物化學而變化,在 Br -存在的情況下發生較高比例的低過電位溶解。在 TFSI-I 電解液的情況下,在低過電勢下只有一個單峰被分解,這表明金屬溶解在低過電勢下的動力學隨著鹵化物尺寸的增加而增強(即 Cl - < Br - ?< I ?- )。
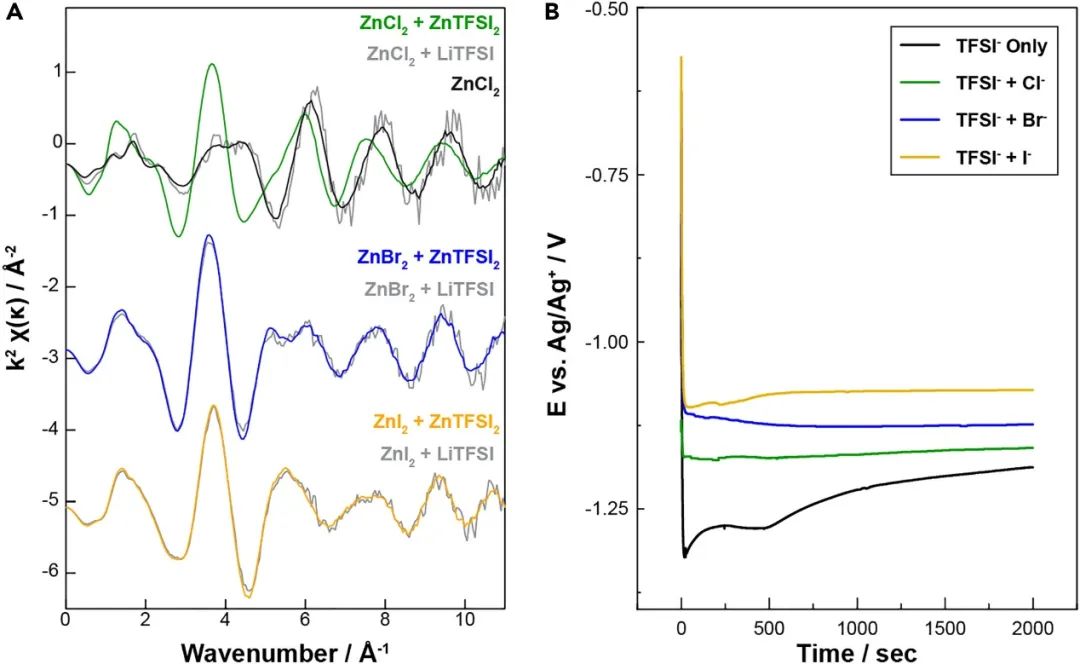
圖4. 陰離子交換的 EXAFS 研究和混合陰離子 CIP 對沉積過電位的影響。
為了評估這種以 Cl 為主的 Zn 2+配位環境在本體溶液中的持久性是否可行, 比較了低濃度 Zn 2+ -鹵化物混合物與過量 TFSI - (5 mM ZnX 2 + 0.2 M LiTFSI) 的 XAS 測量值與濃縮 (0.2 M Zn 2+ ) 混合 TFSI-鹵化物電解液。這些低濃度 Zn 2+電解液直接與電化學研究的濃度方案進行比較。低濃度 Zn 2+電解液的 EXAFS 測量結果顯示在圖 4 A(灰線)與包含先前詳細描述的ZnX 2和 ZnTFSI 2的等效摩爾樣品進行比較在 ZnI 2和 ZnBr 2系統的情況下,低濃度與高濃混合陰離子電解液相同,表明過量的 TFSI -有效地與 Zn-鹵化物(I -和 Br -)絡合物交換形成各種 Zn 2+ –TFSI 絡合物。對于低濃度 ZnCl 2不能得出相同的結論電解液,因為 EXAFS 與高濃度雙陰離子樣品不一致。事實上,低濃度 ZnCl 2電解液的 EXAFS 與高濃度純 ZnCl 2溶液(黑線,圖 4 A)相同,表明游離 TFSI -即使顯著過量也不能輕易交換以產生混合TFSI/Cl -協調環境。這與 Br -和 I -電解液形成鮮明對比,突出了 Zn 2+和 Cl -之間更強的離子配對相互作用. 此外,這些結果支持電化學結果,表明存在有意義的強配位、僅含 Cl 的 Zn 2+配合物群,與含 Br -和 I -的電解液相比,這些配合物需要顯著更高的過電勢才能電化學激活。
綜上所述,這些結果表明,由更弱配位的 Br -和 I -激活的額外溶液物種應該能夠提高混合陰離子電解液中鍍鋅/剝離的速率能力。事實上,來自濃縮電解液(0.2 M 總 Zn 2+ )的循環伏安法(圖 S18)和恒電流電鍍(圖 4 B )均顯示,相對于僅 TFSI,混合陰離子電解液的過電勢顯著改善,過電勢隨著鹵化物締合強度的降低而降低(即,Cl - > Br - > I -), 與圖 3中顯示的稀釋系統的結果直接一致。對這些電解液的沉積物形態進行成像,可以得到一幅關于提高速率對沉積物結構影響的細微差別的圖像。
總結與展望
在這項工作中,作者描述了含有TFSI?和各種鹵素陰離子的雙陰離子Zn2+有機電解液中的突現溶劑化行為。通過拉曼光譜、DFT計算和Zn K-edge EXAFS的建模,作者發現當電解液中也存在鹵素陰離子(X)時,有大量的Zn–TFSI接觸離子對,其中Zn(TFSI)(X)物種被認為是主要的CIP溶劑化結構。這與純ZnTFSI2電解液形成對比,后者在本體中更喜歡第一殼層二甘醇配位,表明鹵化物的存在誘導TFSI?直接配位Zn2+。使用靜態和旋轉圓盤電化學測量描述的電化學行為表明,陰離子締合強度呈周期性趨勢(即I?<Br?<Cl?),有強有力的證據表明,相對于Br和I配位的Zn2+配合物,完全Cl配位的Zn 2+物種持續存在,并需要顯著更高的過電位才能活化。鹵素系列的XAS測量證實了這一假設,其中過量的TFSI?很容易與Br?和I?交換,但無法破壞Zn–Cl離子對。
完全Cl配位的Zn2+配合物的持久性及其在低過電位下缺乏電化學活性對多陰離子電解液的設計具有重要意義。具體而言,含Cl電解液的可逆性的提高是以可用于金屬沉積的總Zn2+的分數為代價的,從而限制了金屬沉積動力學。這表明,對電解液性能的評估必須超越庫侖效率的測量,并考慮本體和界面溶劑化結構,以及由此產生的金屬沉積/剝離的動力學。總的來說,上述電化學、理論和光譜的綜合分析揭示了可逆電化學的明確設計參數,表明陰離子選擇必須優化電解液的整體可逆性和所得配位絡合物的活性。此外,涌現溶劑化行為的存在表明了電解液設計中的額外自由度,其中由于第二陰離子的存在,名義上弱配位的陰離子可以被誘導參與電解液本體中工作陽離子的第一溶劑化殼。本研究中詳述的相互作用的含義遠遠超出此處研究的特定電解液,精確控制陰離子-陽離子相互作用為電解液的設計提供了參考。
審核編輯:劉清
-
鋰離子電池
+關注
關注
85文章
3243瀏覽量
77791 -
電解液
+關注
關注
10文章
849瀏覽量
23156 -
儲能技術
+關注
關注
4文章
181瀏覽量
14323 -
TFSI
+關注
關注
0文章
3瀏覽量
5030
原文標題:阿貢實驗室Chem發文再談Zn2+配位環境
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
潤和軟件助力實驗室行業智慧化發展
NVIDIA 推出 BioNeMo 開源框架,擴大全球生物制藥和科學行業的數字生物學研究規模
LIMS系統在芯片實驗室中的應用
LIMS實驗室管理平臺的實施步驟
LIMS平臺提升實驗室工作效率的方法
如何選擇合適的LIMS實驗室管理軟件
實驗室信息管理系統 LIMS 優勢
蘋果深圳應用研究實驗室正式運營
友思特“未來視界”趣味實驗室 第2講:中草藥的高光譜成像
福祿克產品在實驗室中的應用
阿斯麥(ASML)與比利時微電子(IMEC)聯合打造的High-NA EUV光刻實驗室正式啟用






 工商網監
工商網監
評論