 高通量測序生物信息學分析
高通量測序生物信息學分析
高通量測序技術產生的DNA序列數據長度較短,而且數據量非常巨大。分析了高通量測序環境下大數據的挑戰和機遇,總結并討論了數據壓縮、宏基因組數據序列拼接、宏基因組數據序列分析方面的算法和工具等研究成果。最后,展望了高通量測序下DNA短讀序列數據研究的發展趨勢。
高通量測序分析
高通量測序,一次性對幾百萬到十億條DNA分子進行并行測序,又稱為下一代測序技術,其使得可對一個物種的轉錄組和基因組進行深入、細致、全貌的分析,所以又被稱為深度測序。主要包括:High-throughput Sequencing,Next Generation Sequencing,Deep Sequencing。
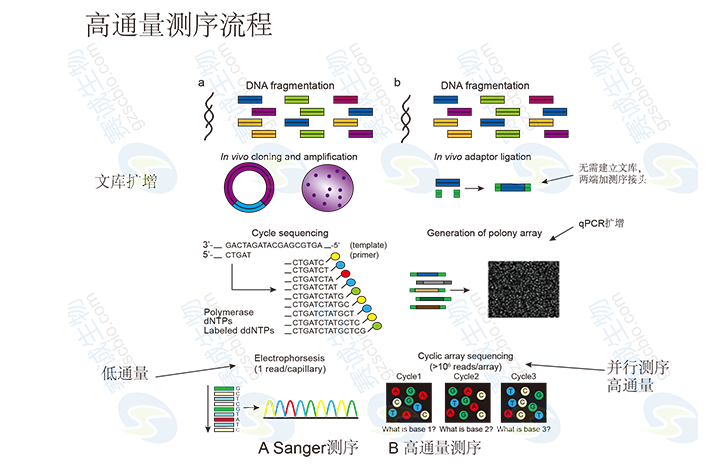
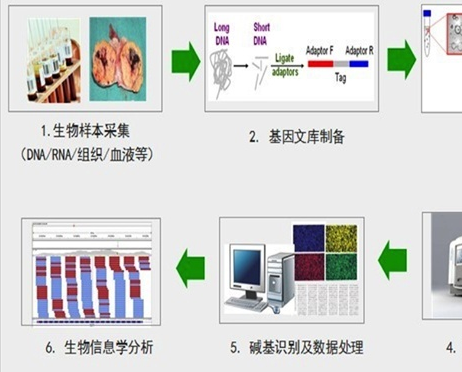
圖1 高通量測序流程
高通量測序應用范圍廣泛:
1 DNA測序:全基因組de novo測序,基因組重測序,宏基因組測序,人類外顯子組捕獲測序。
2 RNA測序:轉錄組測序,小RNA測序,電子表達譜測序。
3 表觀基因組研究:ChIP-Seq,DNA甲基化測序。
基因組測序
基因組測序是對物種的基因組DNA打斷后進行高通量測序,根據是否有已知基因組數據主要分為de novo全基因組測序和基因組重測序。De novo 基因組測序是對未知基因組序列的物種進行基因組從頭測序,利用生物信息學分析手段對序列進行拼接、組裝,從而獲得該物種的基因組圖譜。全基因組重測序是對已知基因組序列的物種進行不同個體的基因組測序,并在此基礎上對個體或群體進行差異性分析。
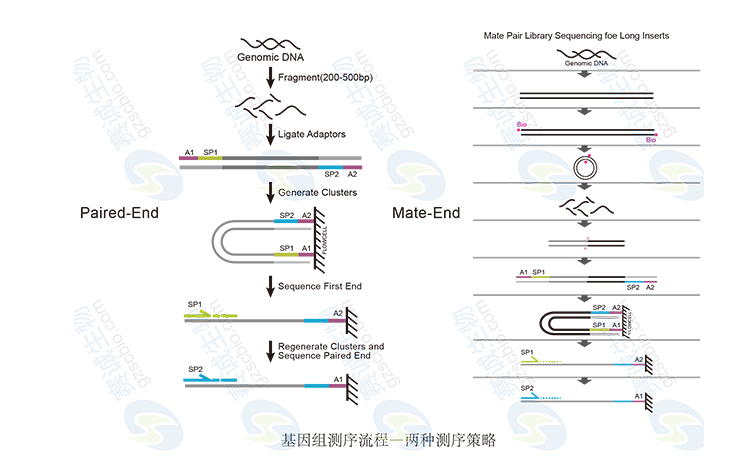
圖2 基因組測序策略
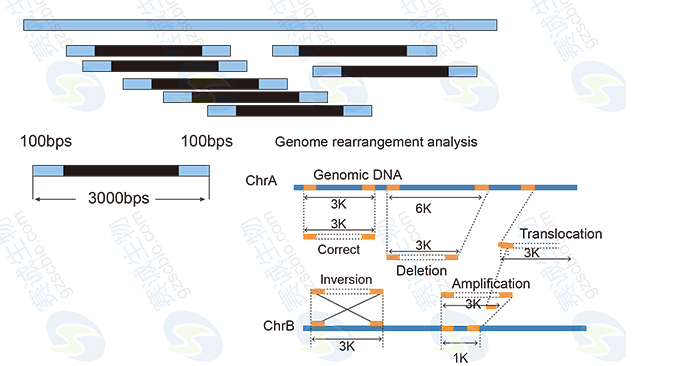
圖3 Paired-end原理
Paired-End方法,基因組打斷后,選擇一定長度(200-500bp)的序列連接兩端接頭進行兩頭測序。Mate-end建庫較復雜,序列打斷后,選取一定長度序列(3-5kb),需先連接生物素,再環化,再打斷,生物素富集,連接兩端接頭進行兩端測序。
基因組測序應用生物信息學分析其結果,主要涵蓋以下幾方面。
1 數據產出處理:圖像識別與Base Calling\去除接頭序列、檢測與去除污染序列等;
2 基因組組裝:原始數據統計、測序深度分析、組裝結果統計等;
3 基因組注釋:Coding Gene注釋、RNA分類注釋、重復序列注釋等;
4 基因功能注釋:GO功能分類、Interpro功能分類等;
5 比較基因組及分子進化分析:SNP/InDel/CNV檢測等。
宏基因組測序
宏基因組測序是對某一特定環境,如腸道、土壤、海水等中的所有微生物進行基因組測序。通過此方法可對該環境中的微生物種類和優勢物種進行檢測,揭示微生物群落多樣性、種群結構、進化關系、功能活性、相互協作關系及與環境之間的關系 。自然環境中很多微生物無法分離培養,而此方法無需對微生物進行分離培養。宏基因組測序方法現在有全基因組的宏基因組測序和16S/18S rRNA宏基因組測序。
1 全基因組的宏基因組測序
通過高通量測序技術,對環境樣品的總 DNA 直接進行全基因組的宏基因組測序,能夠實現微生物群落的物種分類研究、群落結構、系統進化、功能注釋以及物種間的代謝網絡研究,挖掘具有應用價值的基因資源,開發新的微生物活性物質。與傳統的 Sanger法相比,速度快,性價比高,周期短,單個樣品的測序量可以接近飽和。
宏基因組測序信息分析主要包括:拼接組裝,物種分類組成分析,基因預測和功能注釋,生成Profiling table,主成分分析(PCA),篩選與樣品分組顯著相關的因子,多樣品間比較分析等。
2 16S/18S rRNA宏基因組測序
16S/18S rRNA是微生物群落分析和細菌進化研究以及分類研究最常用的靶分子,采用新一代測序技術,對16S/18S rDNA的可變區進行測序分析,不需進行克隆篩選,能全面的反映微生物群體的物種組成,真實的物種分布及豐度信息。
16S/18S rRNA測序信息分析主要包括:物種分類、物種豐度分析,OTU(Operational Taxonomic Units)分析,多樣性分析,系統進化分析,多樣品間的比較分析等。
人類外顯子組捕獲測序
外顯子組是指全部外顯子區域的集合,該區域包含合成蛋白質所需要的重要信息,涵蓋了與個體表型相關的大部分功能性變異。與全基因組重測序相比,外顯子組測序只需針對外顯子區域的DNA,覆蓋度更深、數據準確性更高,更加簡便、經濟、高效。
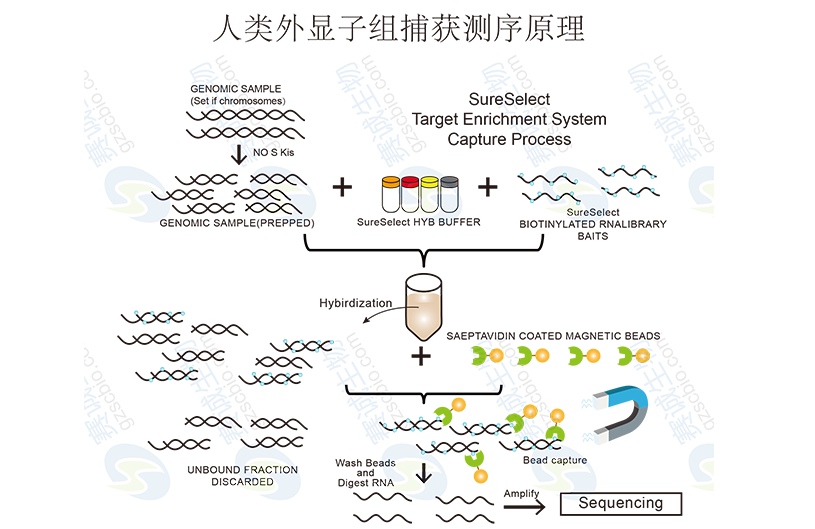
圖4 人類外顯子組捕獲測序原理
外顯子捕獲是指用外顯子芯片雜交,把基因組外顯子序列進行捕獲,然后對所捕獲的序列進行測序。現在常用外顯子芯片有Roche NimbleGen Sequence Capture 2.1M Human Exome Array和Agilent SureSelect Target Enrichment System(Human Exome)。
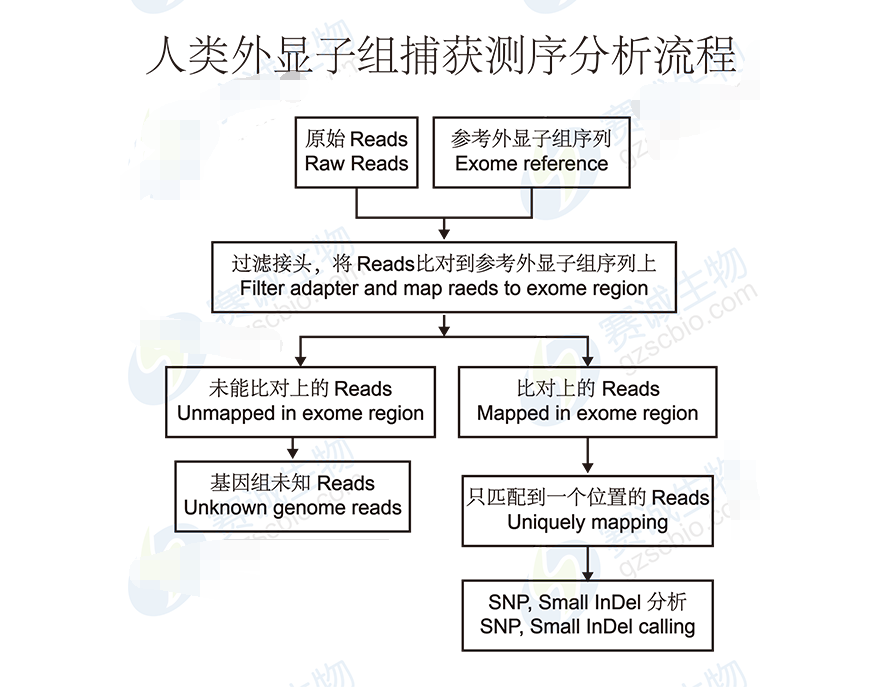
圖5 人類外顯子組捕獲測序分析流程
轉錄組測序
轉錄組即特定細胞在某一功能狀態下所能轉錄出來的所有RNA的總和,包括mRNA和非編碼RNA(Non-coding RNA)。
第二代測序系統可精確檢測單個堿基,并且不受到研究中先驗信息的干擾,科研人員能夠快速地獲得某一物種特定器官或組織在某一狀態下幾乎所有mRNA轉錄本序列,從而能夠開展:UTRs區域界定、可變剪切研究、低豐度新轉錄本發現、融合基因鑒定、cSNP(編碼序列單核苷酸多態性)研究等。
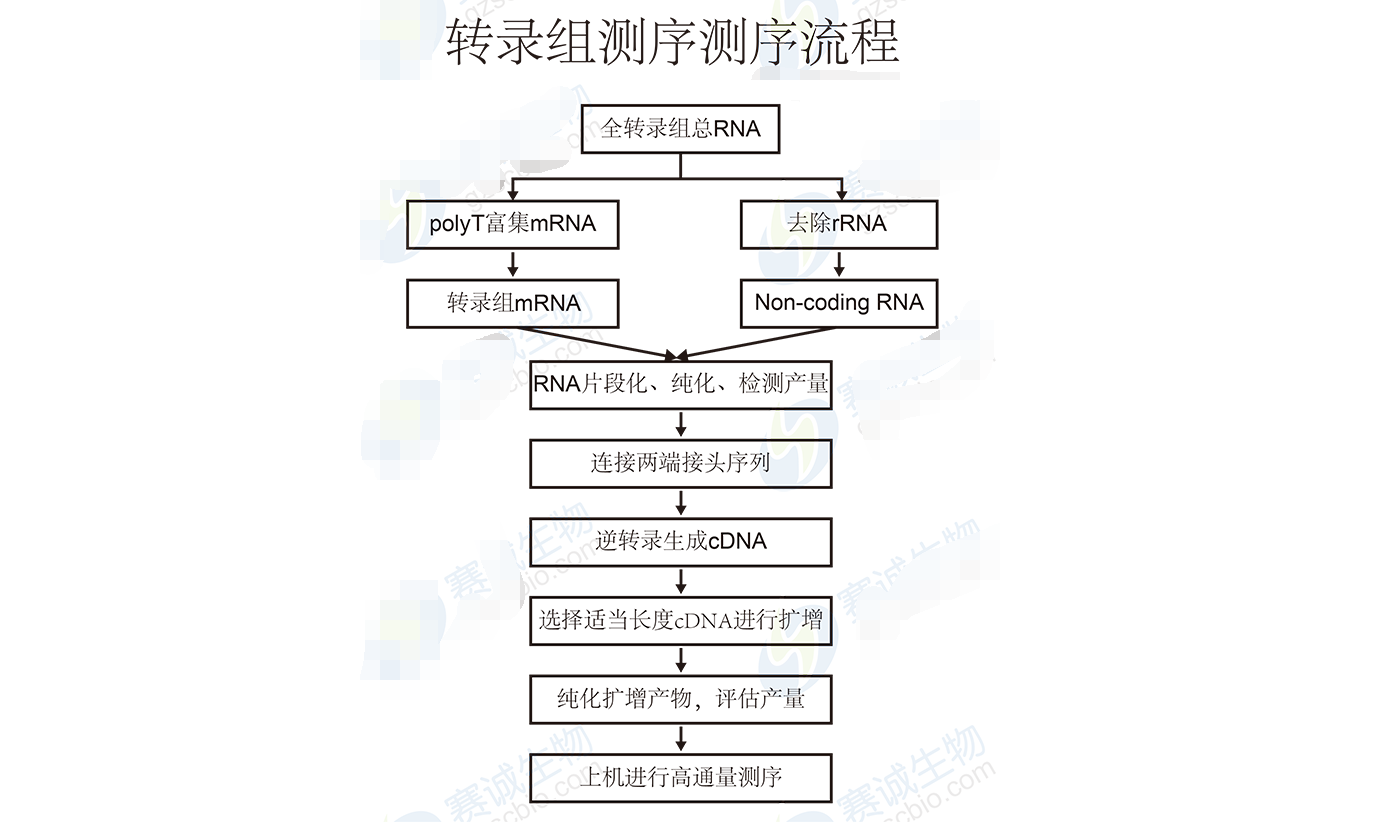
圖6 轉錄組測序流程
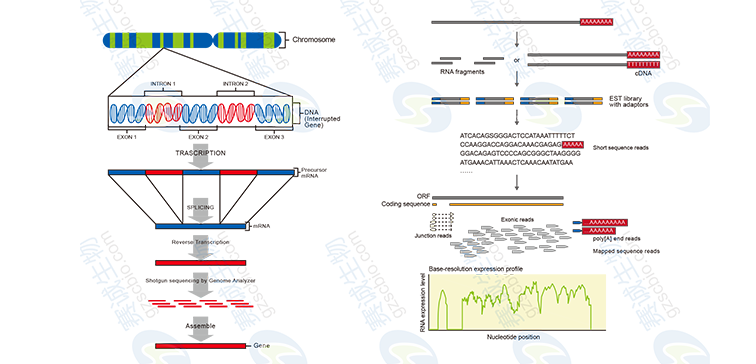
圖7 無參考序列及有參考序列轉錄組測序流程
無參考序列轉錄組分析內容包括:1 測序數據產量統計,數據成分和質量評估;2 Contig及Scaffold長度分布;3 Unigene的長度分布和功能注釋,GO分類,Pathway分析,差異表達分析;4 蛋白功能預測與分類,差異表達基因GO富集和 Pathway富集分析。
有參考序列轉錄組分析內容包括:1 基本數據統計,比對參考序列;2 序列在基因組上在分布;3 測序深度分析、隨機性評估和基因差異表達分析;4 新基因預測,基因可變剪接鑒定和基因融合鑒定等。
電子表達譜測序
電子表達譜測序(Digital Gene Expression, DGE)又稱為基因表達標簽測序(mRNA tag profiling),又稱Tag-SAGE。其原理是通過兩種酶切作用對基因中一段長度為21nt的序列標簽進行測序。由于其測序只針對表達的基因進行測序,產生的數據量相對較小,是研究基因表達譜的經濟而快速的研究手段。是對特定處理條件下的全基因組基因表達譜進行分析,已被廣泛用于功能基因組學和醫學等研究領域。
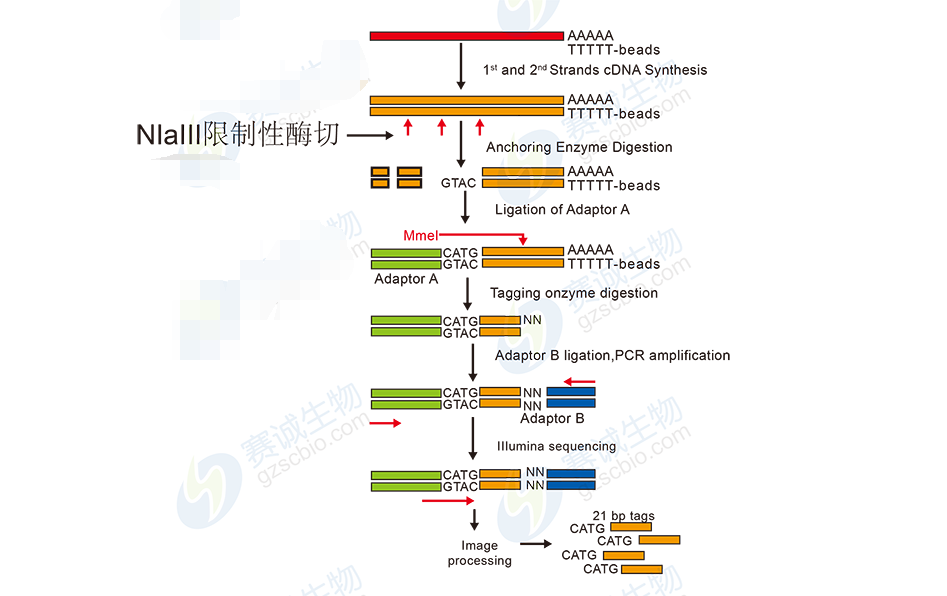
圖8 電子表達譜測序流程圖
電子表達譜分析內容包括:圖像識別與原始堿基數據讀取,去污染、去接頭,標簽序列計數統計,基因組比對與統計,基因序列比對獲得所表達的基因列表,基因差異表達分析,聚類與表達類型分析,GO基因富集與分類分析,Pathway富集與分類分析,蛋白相互作用網絡分析,反義鏈轉錄本與新轉錄本檢測等。
小RNA測序
小 RNA是指長度在21-31nt的內源性非蛋白質編碼RNA,廣泛存在于高等和低等生物體內,其對mRNA的轉錄及轉錄后水平等生命過程起到調節作用。現已知小RNA可歸納成三類:微RNA (miRNA),小干擾RNA(siRNA)和與piwi相互作用的RNA(piRNA)。
miRNA長度為21~24nt,產生于有典型莖環二級結構的原轉錄本(pri-miRNA),在動植物的目標mRNA的降解與抑制方面發揮重要作用。siRNA,長度在19~25nt,產生于長雙鏈RNA,同樣在動植物的目標mRNA的降解與抑制方面發揮重要作用。piRNA,長度26~31nt,由與其相互作用的Piwi蛋白定義,目前研究表明其在配子形成的過程中起作用。
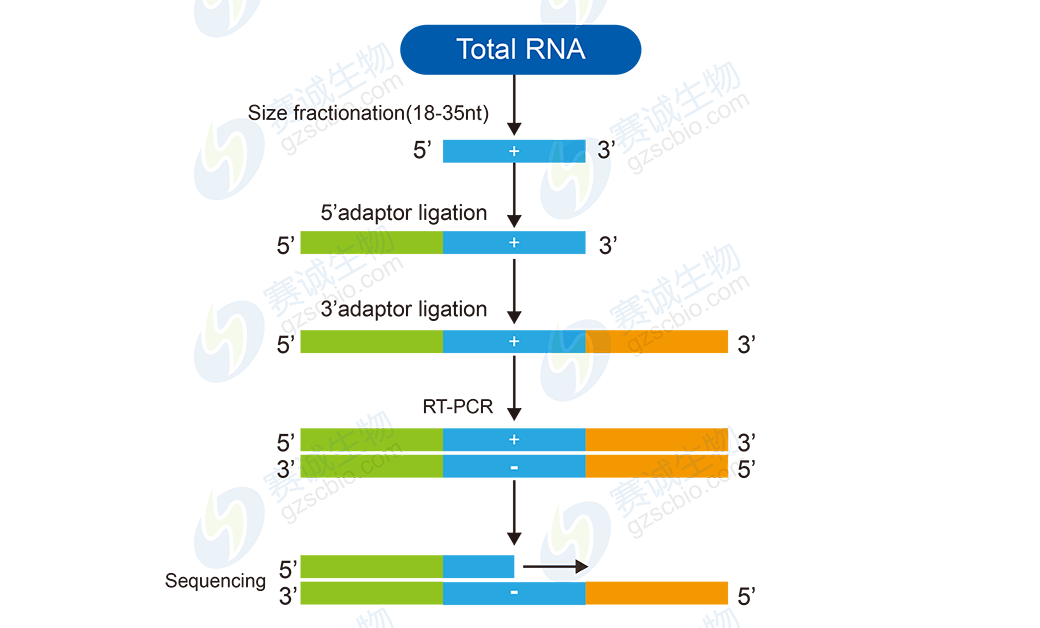
圖9 小RNA測序流程圖
小RNA測序分析內容包括以下兩個主要方面:
1 基本分析:原始數據讀取,去接頭、去污染序列,長度分布統計,基因組比對等。
2 高級分析:Small RNA的分類注釋,miRNA / siRNA / piRNA的鑒定,新miRNA預測,差異表達miRNA聚類分析等。
ChIP-Seq
ChIP-Chromatin Immunoprecipitation染色質免疫共沉淀,是指通過蛋白免疫相互作用,用抗體把和染色質相互作用的蛋白,如組蛋白、轉錄因子等,沉淀下來,從而所獲取與其相結合的DNA序列。ChIP-Seq就是通過高通量測序對ChIP所得到的序列進行測序,從而進行蛋白和DNA相互作用相關研究。
ChIP-Seq分析內容包括:
1 ChIP Sequencing結果與參考基因組序列進行比對。
2 ChIP Sequencing reads 在全基因組的分布:唯一比對reads 在repeats 區域的分布,唯一比對reads 在各基因功能元件上的分布,唯一比對reads 的全基因組覆蓋深度。
3 全基因組peak 掃描:peak 掃描,peak 長度分布統計,peak 的全基因組覆蓋度,peak 在基因功能元件上的分布特征,
4 Peak相關基因分析篩選與GO功能富集分析。
5 多個樣品的差異分析:基于peak 相關基因的差異分析,基于peak 的差異分析。
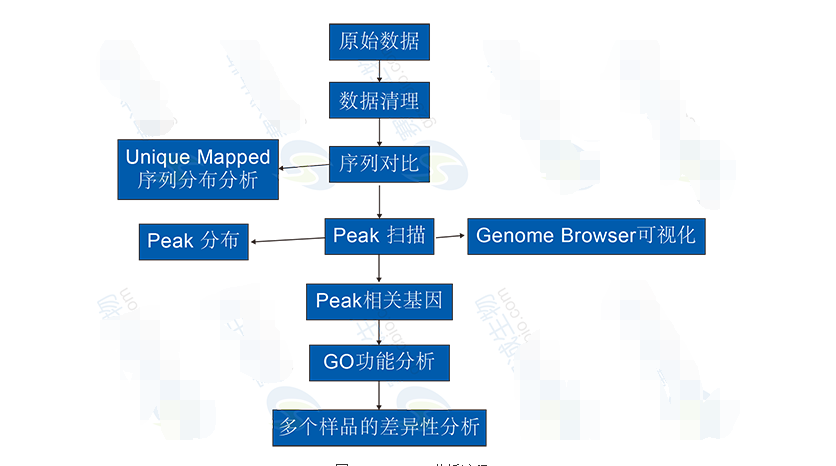
圖10 ChIP-Seq分析流程
DNA甲基化測序
DNA甲基化對機體發育和基因表達有很重要的調控作用,和各種癌癥的發生和發展也有很大相關性,所以對基因組DNA甲基化進行研究是一直來的熱門課題。通過高通量測序來研究DNA甲基化現在主要有兩種方法,一種是MeDIP,是通過與DNA甲基化位點相結合的抗體,進行免疫共沉淀,然后對所得DNA序列進行測序。另一種是Bisulfite Sequencing,是通過Bisulfite處理基因組來區分甲基化位點。
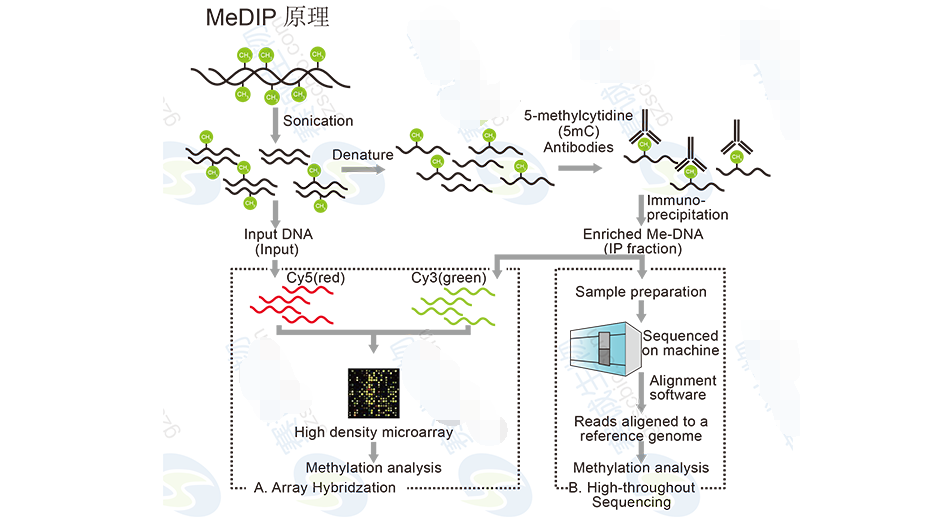
圖11 MeDIP 原理
MeDIP-Seq分析內容包括:
1 MeDIP-seq 序列與參考序列的比對。
2 MeDIP-seq 序列數據在全基因組的分布趨勢: MeDIP-seq 測序reads 在全基因組上每條染色體上的分布,MeDIP-seq 測序reads 在全基因組上的覆蓋深度,MeDIP-Seq 測序reads 在CG、CHG和CHH位點上的覆蓋深度,MeDIP-Seq 測序reads 在不同基因功能元件上的分布,MeDIP-Seq 測序reads 在不同OE含量區域中的分布。
3 統計MeDIP-seq 序列富集區域(peak)的信息:Peak 掃描,Peak 長度數量及比例分布統計,單個樣品Peak 的OE含量分布統計,尋找Peak 相關基因,統計Peak 在不同基因功能元件上的分布。
4 基于Peak 的多樣品間差異分析:分析兩個樣品間的Peak 相關差異基因,對兩個樣品間的差異基因進行GO功能富集分析及pathway 功能分析。
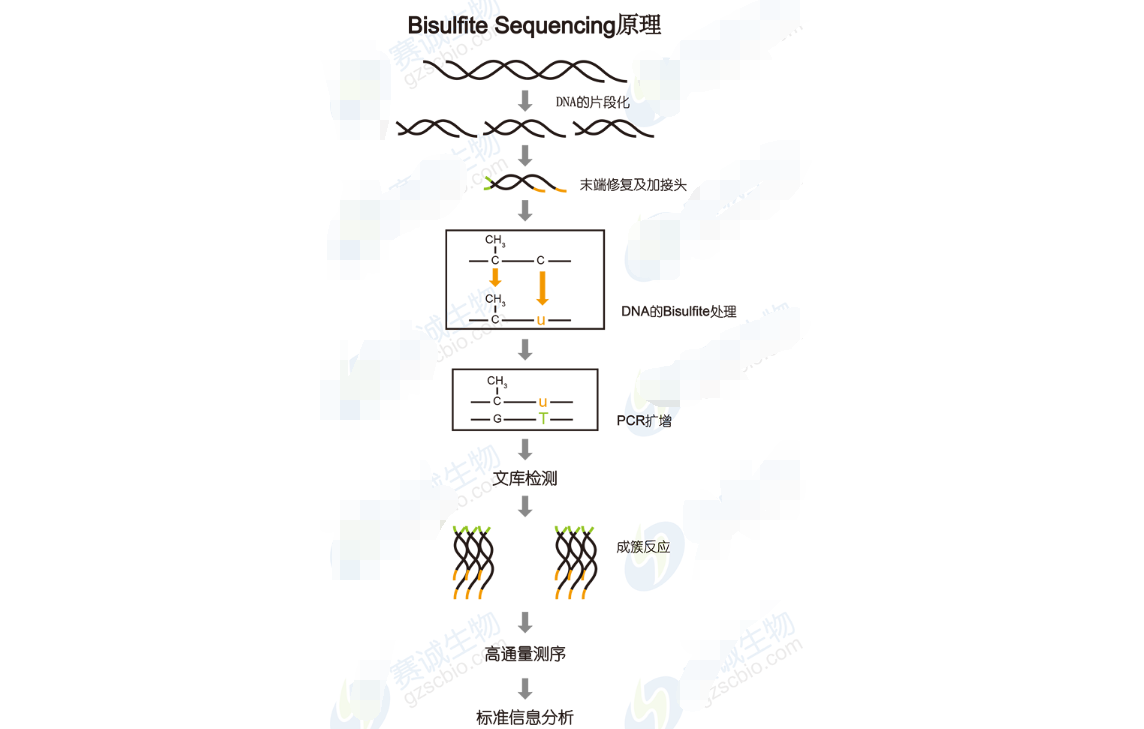
圖12 Bisulfite Sequencing原理
Bisulfite Sequencing分析內容包括:
1 Bisulfite-seq序列與參考序列的比對。
2 深度和覆蓋度分析:C堿基有效測序深度的累積分布,不同reads 測序深度下的基因組覆蓋度。
3 計算C堿基的甲基化水平。
4 全基因組甲基化數據分布趨勢分析:甲基化C堿基中CG, CHG 與CHH的分布比例(H=A、C or T),CG、CHG和CHH中的所有C的甲基化水平,各條染色體中CG、CHG和CHH中C的甲基化水平(該項分析目前只用于“人”),統計不同基因區域內CG、CHG和CHH中C的甲基化水平,不同基因元件區域中CG、CHG和CHH中C的甲基化水平,CHG,CHH中甲基化C附近的9bp序列的序列特征分析。
5 全基因組DNA 甲基化圖譜:染色體水平的甲基化C堿基的密度分布(該項分析目前只用于“人”),Scaffold的甲基化C堿基密度分布(該項分析針對物種:非人),不同基因組區域的甲基化分布特征,基因組不同轉錄元件中的DNA甲基化水平。
6 差異甲基化區域(DMR)分析。
發布評論請先 登錄
相關推薦
高通量測序數據分析:RNA-seq 精選資料分享
全基因組測序的優勢 精選資料分享
高通量測序技術及原理介紹
高通量測序常用名詞匯總
高通量測序技術及其應用
高通量基因測序是什么_高通量測序的意義
Clay Breshears博士討論基因組測序和生物信息學
什么是高通量單細胞RNA測序技術?
廈門大學研發出全新高通量單細胞轉錄組測序方法
披荊斬棘,乘風破浪——真邁生物高通量基因測序儀GenoLab發布
用NVIDIA Clara Parabricks v4.0大眾化和加速基因組測序分析
使用北鯤云在AWS上運行基因分析HPC任務





 工商網監
工商網監
評論