 基因技術被認為是改變未來的技術之一
基因技術被認為是改變未來的技術之一
基因技術被認為是改變未來的技術之一。根據麥肯錫的報告,預計到2025年,全球將會累計產生10億人次的全基因組數據。基因組學所需的數據量如此巨大,用深度學習技術去探索人類基因組密碼便成為了趨勢與未來。本專欄將結合最新的一篇來自于卡耐基梅龍大學的綜述論文,回顧與展望這一交叉學科的發展。
自從2013年變分自動編碼器(VAEs)被提出,2014年Goodfellow提出生成對抗網絡(GANs)起,生成式模型(generative models)深得深度學習研究者的青睞。尤其是當深度學習由于“black box”限制不能充分地推動AI在生物學、基因組學中的發展時,很多學者力求探索生成式模型在其中的應用。
比如,前幾日arXiv上一篇來自斯坦福大學的論文就展現了如何利用 GANs 去編碼可變長度蛋白質的合成 DNA 序列。面對合成生物學這類屬于人類未來的新興學科,人工智能在其中能發揮的巨大作用值得期待。對于想要了解這一領域的學者,本專欄介紹的這篇由卡耐基梅龍大學碩士岳天溦與Eric Xing教授的學生汪浩瀚合著的論文“Deep Learning for Genomics: A Concise Overview”, 綜述了深度學習在基因組學中的應用。文中分析了不同深度模型的優劣勢,舉例講解如何利用深度學習解決基因學問題,并且指出了當前科研所面臨的缺陷和挑戰。

論文鏈接:https://arxiv.org/abs/1802.00810作者GitHub還有一些重要論文的筆記: https://github.com/ThitherShore/DLforGenomics
深度學習應用于基因組學:解密人類遺傳密碼
自從 James D Watson 于1953年將DNA解釋為人類遺傳信息的載體,人們便致力于研究如何更有效地收集生物信息,以及探索由這些遺傳信息主導的生物學過程。于1990年啟動的科學探索巨型工程:人類基因組計劃(Human Genome Project),其宗旨便在于測定組成人類染色體所包含的30億個堿基對組成的核苷酸序列。其目的在于繪制人類基因組圖譜,辨識并破譯其載有的人類遺傳信息。至2001年,人類基因組計劃首次公布了人類基因組工作的草圖。近年來,FANTOM, ENCODE, Roadmap Epigenomics等,以及不同物種的基因組計劃被陸續啟動執行,使得科學家們有更多的途徑和信息去探索基因科技。在這個人工智能技術全面滲透的時代,基因科技作為可以改變人類未來的科技之一,也備受關注。
基因組學不同于傳統的遺傳學,它的數據量非常大。遺傳學研究通常只牽扯到個別基因,但基因組學研究需考慮一個生物體的所有基因,從整體水平上探索全基因組在生命活動中發揮的作用。比如,若對人類基因序列測序,那么信息量級為23對染色體上的30億對堿基排序。
由于基因組學所需信息量巨大,其研究的推動依賴于先進的基因測序技術。Frederick Sanger 發明了測序法后,人類才得以對整個基因組進行測序。DNA微陣列(macroarray)芯片技術的誕生,使得大規模的基因測序成為可能。隨后,2000年首次商用的高通量測序(High-throughput Sequencing, THS)是基因測序領域的一次革命性的技術變革。HTS 可以大規模、低成本、快速地獲得任何生物的基因序列。但 HTS 有一個致命的缺陷,其測序結果是不完整的短序列片段,被稱為讀取單位(reads)。如何高效又精準地拼接這些碎片化的信息,對于HTS一直以來是一種挑戰。近期,一款由Google Brain 聯合 Alphabet旗下公司Verily所開發的開源工具DeepVariant,巧妙地將HTS序列片段的拼接問題轉化為一個圖像處理分類問題。DeepVariant利用了Google Brain 的圖像處理模型Inception,用深度神經網絡來識別HTS測序結果中DNA堿基變異位點,包括基因 組上的單堿基突變(SNP)和小的插入缺失(Indel),從而極大提高了的拼接精度。
另一方面,深度學習模型被廣泛應用于鑒別基因的不同成分,比如外顯子(exons), 內含子( introns), 啟動子(promoters), 增強子(enhancers), positioned nucleosomes, 剪接位點( splice sites), 非轉錄區 (untranslated region, UTR)等。同時,有豐富的數據種類可被用于基因組學的研究:基因微列陣(microarray),RNA-seq expression,轉錄因子(DNA結合),轉錄后修飾(RNA結合),組蛋白修飾(histone modifications)等。許多信息門戶比如GDC, dbGaP, GEO都為廣大科研工作者提供了這類數據來源。
面對日益精進的生物技術,和飛速發展的深度學習與人工智能技術,用深度學習去探索人類基因組密碼便成為了趨勢與未來。這篇paper分析了不同深度模型的優劣勢,并站在不同生物問題的角度,談及深度學習在其中的應用。文末指出了當前科研工作的一些缺陷和挑戰。
深度學習模型對比:CNN、RNN、自動編碼器、新興模型結構
深度學習發展至今,CNN, RNN, 前饋神經網絡(feed-forward neural networks),自動編碼器(Auto-Encoders)等種類繁多。在實際應用中,如何利用各類模型的優勢去解決不同類型的基因學問題呢?
CNN
近幾年,CNN在計算機視覺領域取得了空前的成功,這得益于其擅長的捕捉空間信息特征的能力。CNN在圖像處理領域卓越的性能亦可被用于基因組學研究中。類比于有R, G, B三個顏色通道的二維圖像,基因序列的一個窗口可以被看做有4個頻道(A, T, C, G)的一維序列,由此便可通過一維卷積核進行單序列分析(single sequence assays)。CNN能夠逐步提取圖像特征的能力,可以被用來鑒別基因圖像中有意義的圖形,從而應用于 motif identification 和 binding classification 等問題中。
RNN
RNN擅長于處理序列性數據,故而成功應用于自然語言處理領域。由于基于序列很長,且位點之間有復雜的相關性,故RNN類結構(LSTM, bi-LSTM, GRU)也被很多基因組學研究者青睞,應用于通過基因序列的信息研究非編碼DNA(non-coding DNA)功能,或進行亞細胞定位( subcellular localization)等。
Auto encoders
自動編碼器是一個由來已久的神經網絡模型,以往常被用于初始化神經網絡參數。在近年VAE的思路提出后,不少學者又開始應用VAE或Autoencoders類(Contractive Autoencoders, Stacked Denoising Autoencoders, Denoising Autoencoders)模型來進行數據降維,或試圖借此捕捉基因序列間隱含的依賴關系。
新興模型結構
由于基因組數據量大,生物體各部分間依賴關系復雜,單一形式的深度神經網絡模型已經不能滿足人們對效率和精度的高要求,目前在基因組研究中取得突破性成功項目,都運用、結合了多個深度學習網絡模塊。比較常見的幾種方式包括:
CNN+RNN結構,利用CNN初步處理DNA序列局部特征,后結合RNN挖掘DNA序列間的依賴性,比如DanQ(下圖),在輸入層將DNA序列表示成one-hot編碼,分別經過卷積層和池化層后,用LSTM進行進一步特征提取;
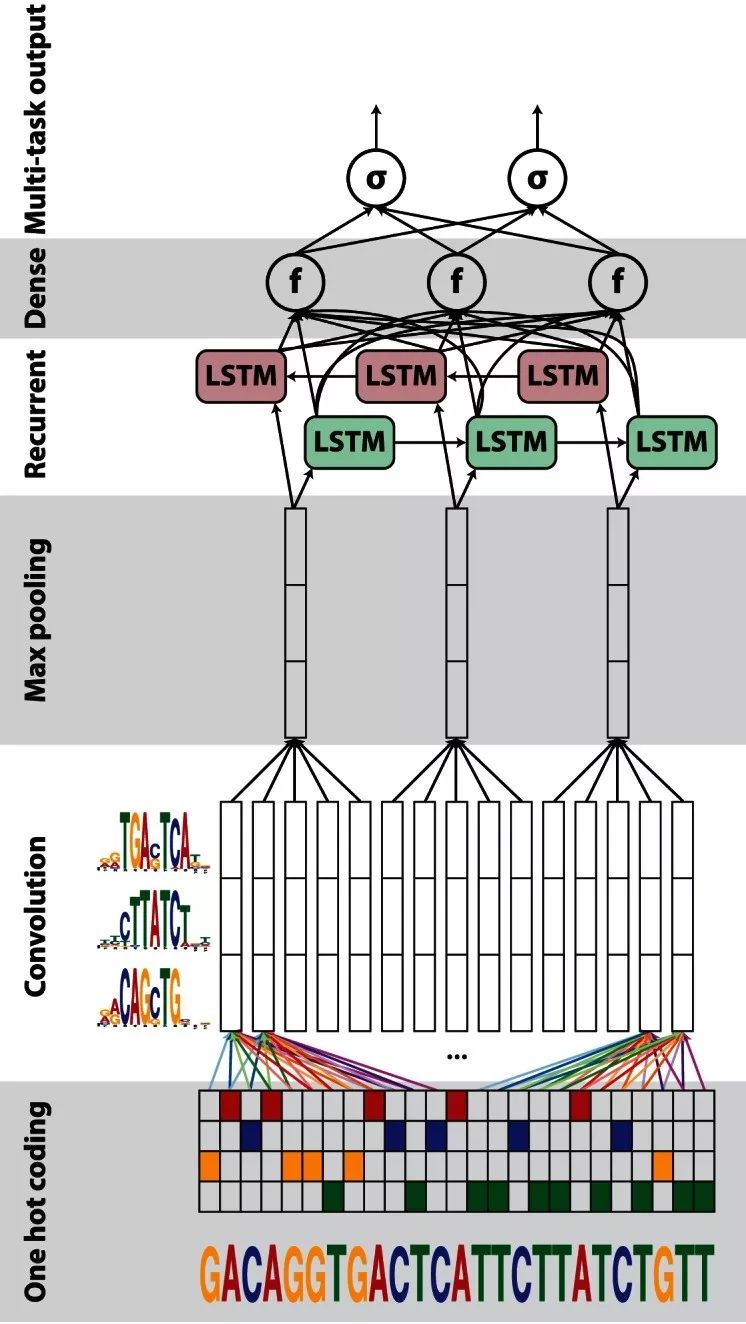
堆疊的(stacked)網絡結構,利用多層網絡去捕捉深層次的相互依賴關系,比如 DST-NNs;
同一網絡結構的并行運用,比如DeepCpG,將兩個CNN各自作為整體模型的兩個子模塊(sub modules),分別從CpG sites和DNA序列提取特征,并在高層模塊(Fusion Module)融合這兩部分信息;
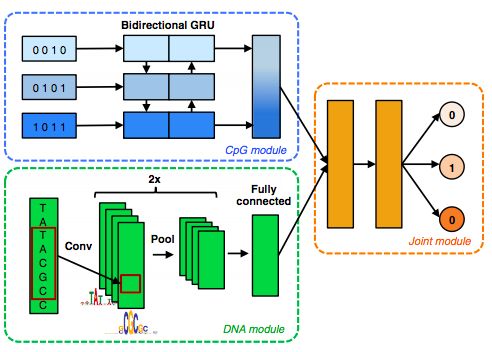
對于這些新興的,更復雜的網絡結構,雖然其應用效果優于傳統統計或機器學習,但其泛化性,可解釋性還亟待探究。
深度學習模型的可解釋性和建模方式
模型可解釋性
深度學習“黑箱”是人們一直在力求改進的一個缺陷。由于深度學習方法本身的這點不足,人們在直接將其應用在基因組學中,力求解釋基因問題時,希望能夠賦予自己的模型適當的可解釋性。作者介紹了一些經典的計算機視覺領域對CNN的解釋,和基因組應用中人們結合問題對深度學習模型解釋的例子。比如可視化CNN各層提取的特征,或采用saliency map,又比如 Deep GDashboard 模型,它探索比較了CNN和RNN各自在同一個問題中發揮的性能。
建模方式討論
想要提高深度學習在基因組學中應用的效果,除了提升模型結構上的設計,還可以考慮從模型訓練上提高。由于基因組數據量之大,完整訓練一個精準有效的網絡耗時且困難,所以可以考慮遷移學習(transfer learning)。很將某個訓練好的模型(部分或整體)用作另一個問題的初始化,或用已有模型直接進行特征提取分析。這種思路在計算機視覺領域早已應用。此外,可以考慮同時解決兩個或多個相關的問題(多任務學習, multitask learning),在建模中利用他們共有的信息成分。考慮到基因組數據的多樣性,可以考慮multi-view learning,建立模型利用該問題的不同數據類型。這可以通過concatenating features, ensemble methods, or multi-modal learning (為不同模塊/不同數據類型設計相應的sub-networks,并在網絡高層結構中融合各個子網絡的信息) 來實現。
深度學習在基因組學問題中的應用
論文中回顧了深度學習在以下這些領域中的應用,并詳細介紹了一些近年的值得矚目的研究:
1. 基因表達(gene expression):特征和預測2. 調控基因組學(regulatory genomics):
啟動子(promoters)和增強子(enhancers)
Functional Activities
Splicing
轉錄因子(Transcription Factors) and RNA-binding Proteins
亞細胞定位(Subcellular Localization)
突變(Mutations) and Variant Calling
3. 結構基因組學(structural genomics):
蛋白質的結構分類(Structural Classification of Proteins)
蛋白質二級結構(Protein Secondary Structure)
Contact Map
挑戰和展望
想要建立深度學習模型解決基因組學問題,需要明確現有一些限制和挑戰,才能更有全局觀,更有目的性的開發更有效的模型。
數據局限性
獲取生物學數據通常耗財耗時,尤其是當我們想通過基因組學數據研究某種稀有性狀/疾病時,數據來源十分匱乏。
作者介紹了以下幾種情況下應對數據所帶來的局限性的一些對策和論文:1.數據各類之間不平衡(class-imbalanced)或部分數據沒有標簽(labels)
2.數據類型不同(Various Data Sources)3.數據來源混雜(Heterogeneity and Confounding Correlations):heterogeneous datasets是醫療數據中很常見的問題。人種的不同,人群的區域性,數據采集的不同批次,都會造成一些誤導因素(confoundering factors)需要模型去處理。
特征提取
在應用中,很多時候我們會采用一些人工提取的特征(hand-engineered features),但這通常需要相應領域的專家協助。雖然譬如CNN這樣的模型,可以有效地提取數據中的特征,但這對模型的設計和調參要求較高。故若有好的特征提取方式,可以有效加速模型訓練,推動科研進程。作者談及了幾種基于拓撲學(topology)的特征提取方式,和一些特征表示方式。
如下圖,這是一個利用了拓撲學中持續同調(persistent homolgy)概念提取蛋白質三維結構中特征的思路。作者從蛋白質出發建單純復形(simplicial complex),從其中拓撲不變量提取特征,并成功地應用于包括蛋白質superfamily分類,protein-ligand binding等多個問題中。
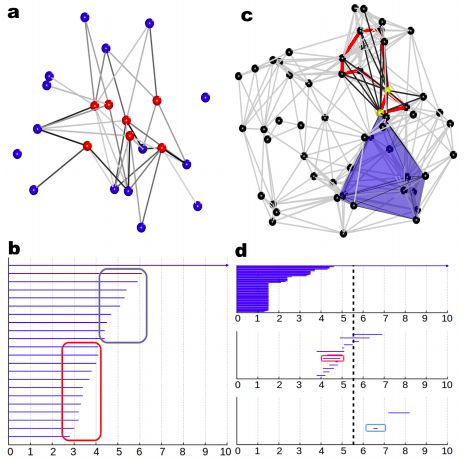
前文討論過各種模型的優劣勢,故而在設計模型時,我們應根據問題選擇合理的設計。同時,也可以在模型參數中引入一些生物學背景知識(prior information),在有限的數據下,盡可能有效地利用現有的信息。
最后,想要讓深度學習在基因組學研究中發揮巨大的作用,我們還有很長的路要走。從生物科技上客服獲取數據的困難,從深度學習方面貼合特定問題開發合適的模型。我們應謹記現有的困難和挑戰,繼續推動這個學科的發展。
-
深度學習
+關注
關注
73文章
5511瀏覽量
121386
原文標題:深度學習 + 基因組學:破譯人類 30 億堿基對
文章出處:【微信號:AI_era,微信公眾號:新智元】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
Micro-LED技術解析
ARM技術是什么?國內有哪些ARM廠家呢?一起來了解一下!
30s高能速遞 | 第三屆 OpenHarmony技術大會精彩搶鮮看
單片機為什么被認為是一門簡單的技術?

碳化硅器件的應用領域和技術挑戰
3個技巧揭秘:開合屏技術如何改變未來!
干貨 | 一文讀懂國內外傳感器技術及差距
多尺度浸入式3D打印策略,用于人體組織和器官的精準制造
一文詳解超算中的InfiniBand網絡、HDR與IB






 工商網監
工商網監
評論