 電子發(fā)燒友App
電子發(fā)燒友App


【研究背景】
鈉離子電池(SIBs)作為有前途的儲(chǔ)能設(shè)備之一,在過(guò)去幾年中引起了廣泛的興趣,因?yàn)殁c的成本低,操作原理與鋰離子電池(LIBs)相似,以及Na+離子的獨(dú)特物理化學(xué)特性。在眾多的候選陽(yáng)極材料中,錫(Sn)特別有吸引力,因?yàn)樗哂懈呃碚撊萘浚?47 mAh g-1)、大體積密度(7.28 g cm-3)和低工作電壓(0.3-0.4 V)。
然而,Na+的大離子半徑和重離子質(zhì)量不僅在循環(huán)時(shí)引起巨大的體積變化(~420%),而且還延遲了電荷在體積中的傳輸。因此,固體電解質(zhì)界面(SEI)的反復(fù)形成、不可控的粒子粉碎以及循環(huán)過(guò)程中的電極開(kāi)裂/脫落,導(dǎo)致低庫(kù)侖效率和快速的容量衰減。
為了解決這些問(wèn)題,人們開(kāi)發(fā)了多種策略,如尺寸控制到納米,與碳材料的均勻復(fù)合,截止電壓的精細(xì)控制,等等。例如,廣泛報(bào)道的Sn/C納米復(fù)合材料總是涉及費(fèi)力的制備、復(fù)雜的儀器和昂貴的試劑。此外,納米復(fù)合材料中的碳材料不僅降低了比容量,而且還降低了攻絲密度和初始庫(kù)倫效率。
因此,有必要尋求新的方法來(lái)改進(jìn)Sn陽(yáng)極。使用微小的錫顆粒(μ-Sn)作為陽(yáng)極材料可以有效地緩解對(duì)納米材料的制備成本、壓實(shí)密度和初始庫(kù)侖效率的擔(dān)憂。但如何保持其穩(wěn)定的循環(huán)是社會(huì)的一個(gè)巨大挑戰(zhàn)。到目前為止,針對(duì)μ-Sn設(shè)計(jì)的策略還很少報(bào)道。
【工作介紹】
本工作將K+離子引入到醚基電解質(zhì)中。然后,K+和Na+離子被電場(chǎng)驅(qū)動(dòng)到電極表面。但是K+離子不會(huì)被還原或納入μSn中,因?yàn)镹ernst方程、分子動(dòng)力學(xué)(MD)模擬和密度泛函理論(DFT)計(jì)算估計(jì)的氧化還原電位低,解離能量大。K+離子會(huì)聚集在 "熱點(diǎn) "上,并通過(guò)靜電屏蔽作用減緩局部的鈉化,促進(jìn)均勻的電化學(xué)反應(yīng)。它增強(qiáng)了電極的穩(wěn)定性,正如化學(xué)機(jī)械模型和掃描電子顯微鏡(SEM)圖像所證明的。
因此,該電極在2 A g-1的條件下經(jīng)過(guò)3000次循環(huán)后保持了565 mAh g-1的高比容量,比電解質(zhì)中沒(méi)有K+的情況好很多。在全電池和μ-Bi中也證明了K+離子的改進(jìn)性能。類似的概念已經(jīng)應(yīng)用于金屬鋰陽(yáng)極,但據(jù)我們所知,還沒(méi)有應(yīng)用于合金型陽(yáng)極。更重要的是,這種方法不涉及費(fèi)力的制備、復(fù)雜的儀器和昂貴的試劑,因此顯示出對(duì)其他SIB陽(yáng)極材料的潛力。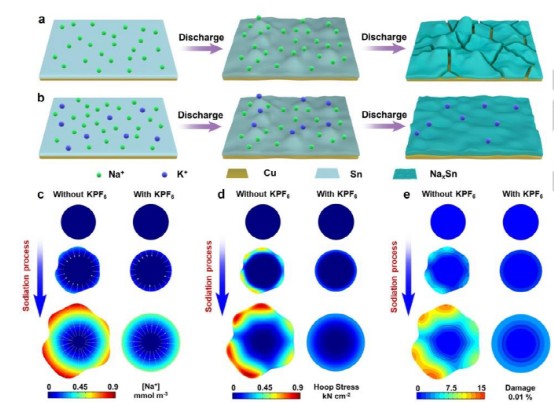
圖1、K+離子的靜電屏蔽作用,增強(qiáng)了電極的穩(wěn)定性。
(a, b) 合金型陽(yáng)極(a)沒(méi)有或(b)在電解質(zhì)中加入K+的結(jié)構(gòu)演變。(c)Na濃度、(d)環(huán)向應(yīng)力和(e)在沒(méi)有或有電解液中的K+的情況下,在鈉化過(guò)程中對(duì)Sn的結(jié)構(gòu)破壞的化學(xué)-機(jī)械模型。 圖1說(shuō)明了K+對(duì)增強(qiáng)電化學(xué)性能的靜電屏蔽作用。由于活性材料、聚合物粘合劑和導(dǎo)電碳在電極中的不均勻分布,Na+在特定部位的局部富集和優(yōu)先鈉化幾乎是不可避免的(圖1a)。局部鈉化的進(jìn)展將引起內(nèi)在應(yīng)力的不均勻分布,并最終導(dǎo)致電極破裂。
相比之下,添加K+將有效地緩解上述負(fù)面影響。如上所述,在這種情況下,K+比Na+更難被還原。因此,當(dāng)K+被電場(chǎng)驅(qū)動(dòng)到活性材料附近時(shí),它不會(huì)被還原,而是優(yōu)先吸附在Na+離子應(yīng)該占據(jù)的熱點(diǎn)處(圖1b)。 K+的持續(xù)積累將呈現(xiàn)出一個(gè)內(nèi)在的電場(chǎng),并抵消了Na+到達(dá)這些位置的驅(qū)動(dòng)力,這被稱為典型的靜電屏蔽。
然后,在熱點(diǎn)位置的局部鈉化將放緩,有利于均勻的體積膨脹并改善電極的穩(wěn)定性。這個(gè)結(jié)論也得到了環(huán)向應(yīng)力和損傷分析的支持。為了簡(jiǎn)化計(jì)算,只有活性材料(μ-Sn),而不是整個(gè)電極,被考慮到模擬中,因?yàn)樗鼈冊(cè)陔姌O中占主導(dǎo)地位。
此外,它們的體積變化是結(jié)構(gòu)應(yīng)力的主要來(lái)源。盡管μ-Sn經(jīng)歷了復(fù)雜的相變,但在這些相變過(guò)程中結(jié)構(gòu)應(yīng)力的演變是相似的。因此,為了清楚起見(jiàn),本化學(xué)機(jī)械模型只使用了其中一個(gè)相變,即Sn/NaSn3。圖1c-1e顯示了在這一轉(zhuǎn)變過(guò)程中Na濃度、環(huán)向應(yīng)力和μ-Sn的結(jié)構(gòu)損傷的演變。
在一個(gè)典型的過(guò)程中,由于內(nèi)部體積膨脹,鈉的外殼逐漸向外擠壓,導(dǎo)致粒子表面產(chǎn)生拉伸箍應(yīng)力。拉伸應(yīng)力隨著鈉化過(guò)程而逐漸累積。當(dāng)拉應(yīng)力超過(guò)NaxSn的閾值時(shí),顆粒會(huì)變形或斷裂以釋放應(yīng)力。在沒(méi)有K+的情況下,由于各向異性的形態(tài)、粗糙的顆粒表面和電極中不均勻的材料分布,Na+通量容易誘發(fā)不均勻的鈉化過(guò)程和μ-Sn的各向異性膨脹(圖1c)。
然后,表面的拉應(yīng)力加劇,增加了潛在的粒子損傷(圖1d和1e)。相反,K+通過(guò)靜電屏蔽使粒子表面的Na+分布正常化,促進(jìn)了均勻膨脹(圖1c)。然后,相鄰材料網(wǎng)格的拉應(yīng)力相互抵消,導(dǎo)致較低的拉應(yīng)力和較少的顆粒損壞(圖1d和1e)。這些結(jié)果支持K+的加入有可能促進(jìn)均勻的鈉化和減少顆粒的損傷。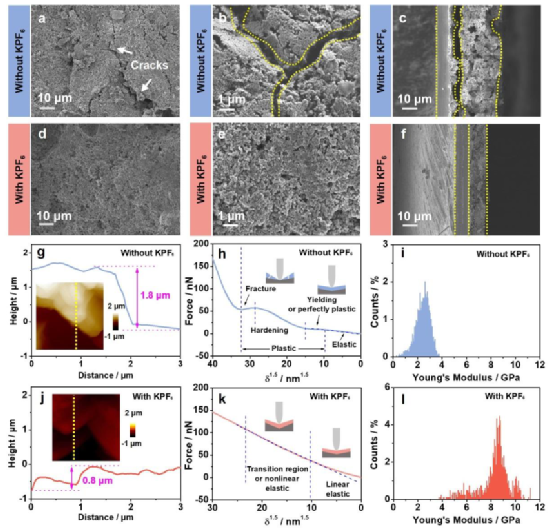
圖2、通過(guò)KPF6改善電極的穩(wěn)定性和機(jī)械性能。
(a-f) 電解液中沒(méi)有K+或(d-f)30個(gè)周期后的電極的SEM圖像。(g, j) AFM圖像(插圖)及其相應(yīng)的高度曲線,(h, k) 力的反應(yīng)及其相應(yīng)的卡通插圖(插圖),(i, l) 分別在沒(méi)有或在電解質(zhì)中加入K+后的電極的楊氏模量分布。
在沒(méi)有K+的電解液中循環(huán)30次后,在電極表面很容易看到裂縫和斷裂(圖2a和2b),表明循環(huán)后的電極穩(wěn)定性較差。同時(shí),電極材料變得松散并從集流體上脫落(圖2c)。結(jié)果表明,巨大的應(yīng)力對(duì)電極造成了明顯的損害,這增加了電荷傳輸電阻,導(dǎo)致容量下降。
相反,在電解液中含有K+的情況下,基本上保持了電極的完整性(圖2d和2e)。電極表面沒(méi)有明顯的裂縫。電極材料仍然保持致密并牢固地附著在集流體上(圖2f)。這些結(jié)果證實(shí)了電極穩(wěn)定性的增強(qiáng),與上述理論預(yù)測(cè)的結(jié)果非常吻合。
AFM得出類似的結(jié)論。圖2g顯示了在電解液中沒(méi)有K+的情況下,3個(gè)循環(huán)后電極的高度曲線和表面形貌,電極表面是粗糙的。表面上的臺(tái)階和/或山脊會(huì)強(qiáng)化電場(chǎng),豐富局部的Na+濃度,從而加速局部生長(zhǎng),增加應(yīng)變/應(yīng)力。然后,電極材料變形,在反復(fù)循環(huán)后出現(xiàn)裂紋。
而在電解液中含有K+的電極循環(huán)是相對(duì)光滑的(圖2j),與循環(huán)前的情況相似。這種良好的穩(wěn)定性也可能與該電極對(duì)結(jié)構(gòu)應(yīng)力的反應(yīng)有關(guān)。如圖2h所示,在沒(méi)有K+的情況下循環(huán)的電極的力的曲線顯示了電極破裂的典型特征,即力隨著壓痕深度的增加而下降。
它符合SEM圖像所觀察到的情況。然而,這種獨(dú)特的特征并沒(méi)有出現(xiàn)在電解液中含有K+的電極循環(huán)的力的曲線上(圖2k),這表明在這種情況下對(duì)力有良好的抵抗力。這方面的見(jiàn)解可能與μ-Sn的均勻膨脹有關(guān),它使內(nèi)在應(yīng)力在一定程度上被抵消。因此,電極對(duì)本征應(yīng)力的耐受性更好,并表現(xiàn)出卓越的穩(wěn)定性。這一結(jié)論直接得到了楊氏模量的支持。無(wú)K+循環(huán)的電極的楊氏模量只有2.7GPa,比有K+的8.3GPa小很多(圖2i和2l)。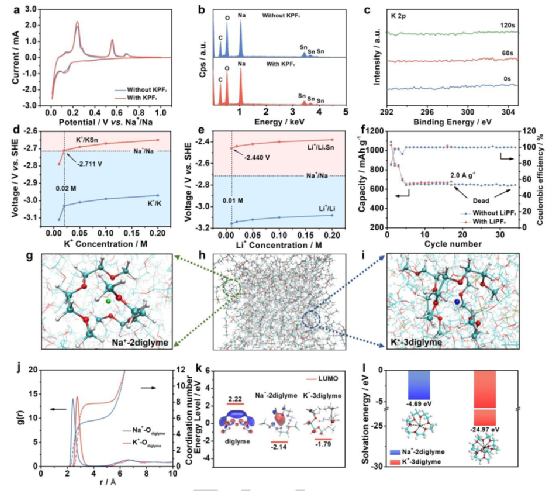
圖3、K+離子不參與μ-Sn的合金化/焊接反應(yīng)。
(a) 電解液中含有或不含有K+離子的電極循環(huán)的CV曲線,(b) EDS光譜和(c) XPS光譜。(d) K+/K和K+/KxSn的氧化還原電位隨K+離子濃度的變化。(e) Li+/Li和Li+/LixSn的氧化還原電位隨Li+離子濃度的變化。(f) 電解液中有無(wú)Li+的Sn||Na的循環(huán)性能,(g, h, i) 電解液中Na+和K+的典型溶解結(jié)構(gòu)快照,(j) RDF曲線,(k) LUMO的能級(jí),(l) Na+-2diglyme和K+-3diglyme的溶劑化能量。(k)中的插圖顯示了分子軌道的等值面,(l)中的插圖描述了相應(yīng)的溶劑化結(jié)構(gòu)。
K+對(duì)電化學(xué)性能的另一個(gè)可能影響是在循環(huán)過(guò)程中與μ-Sn合金化。為了澄清這一點(diǎn),在循環(huán)的電極上測(cè)量了CV曲線、LSV(線性掃描伏安法)曲線、能量色散光譜(EDS)和X射線光電子光譜(XPS)。如圖3a所示,在電解質(zhì)中含有或不含有K+的電極循環(huán)顯示出相同的陰/陽(yáng)極峰,表明K+對(duì)電化學(xué)反應(yīng)的影響可忽略不計(jì)。
換句話說(shuō),K+不參與合金化/去合金反應(yīng)。這一結(jié)論也被EDS和XPS所證實(shí)。圖3b顯示了在電解液中含有或不含有K+的完全放電電極的EDS光譜。無(wú)論是否加入K+,在這兩個(gè)電極中都沒(méi)有檢測(cè)到K元素在3.31 keV (Kα)和3.59 keV (Kβ)的明顯信號(hào)。
這些結(jié)果排除了K參與合金化/去合金反應(yīng)的可能性。同時(shí),在用K+循環(huán)的電極中Na/Sn的原子比是~3.84,接近Na15Sn4的數(shù)據(jù)(3.75),該數(shù)據(jù)被公認(rèn)為是Sn的完全放電產(chǎn)物。這一結(jié)果表明,K+提高了電極中Sn的利用率,因?yàn)樗鼫p少了Na+在熱點(diǎn)上的吸附,使Na+可以在其他位置上進(jìn)行電化學(xué)合金化。
電極中沒(méi)有K+也被不同深度的XPS光譜所證明(圖3c),沒(méi)有觀察到K的可見(jiàn)峰。K+的加入并不直接改變電極表面的固體-電解質(zhì)-中界面(SEI)膜。因此,在兩種情況下,Rct的差異很小。這一結(jié)果也得到了LSV的支持。電解液中含有或不含有K+的電池顯示出類似的曲線,證實(shí)了K+物種沒(méi)有被還原。
為了在理論上理解這一結(jié)果,詳細(xì)討論K+/K(Na+/Na)的實(shí)際氧化還原電位和我們案例中K+/KxSn的合金化電位是非常必要的。眾所周知,K+/K的標(biāo)準(zhǔn)氧化還原電位(-2.931 V vs. SHE)低于Na+/K的氧化還原電位。K+/K的實(shí)際氧化還原電位會(huì)隨著K+的低濃度而進(jìn)一步降低(藍(lán)線,圖3d)。
在本例中,K+的濃度為0.02M,導(dǎo)致氧化還原電位為-3.031V( vs. SHE),使得K+/K的電化學(xué)還原更加困難。然而,K+有可能與Sn進(jìn)行電化學(xué)合金化,在~0.32 V(vs. K+/K)開(kāi)始。如果參考電極被歸一化為SHE,K+/KxSn的起始電位將是-2.611 V(vs. SHE)。
然后,在本案例中,低濃度被考慮在內(nèi)。K+/KxSn的氧化還原電位被估計(jì)為-2.711 V(vs. SHE,紅線,圖3d),幾乎與Na+/Na的相同。考慮到電極過(guò)電位,K+/KxSn的氧化還原反應(yīng)必須發(fā)生在這個(gè)電位以下。這意味著,如果截止電壓被控制在>0.01V(vs. Na+/Na),KxSn的形成將被抑制。這些結(jié)果解釋了為什么K+不參與SIBs中Sn的電化學(xué)反應(yīng)。
然而,如果把K+換成Li+,情況就完全不同了。因?yàn)長(zhǎng)i+/LixSn的起始電位是-2.44V(vs.? SHE,圖3e),Li+有可能在鈉化過(guò)程中與Sn合金化。這將使有限的Li+的消耗,從而使電化學(xué)性能不能與K+相比(圖3f)。因此,選擇一個(gè)合適的陽(yáng)離子進(jìn)行靜電屏蔽是至關(guān)重要的。
基于Nernst方程的討論是簡(jiǎn)化的,因?yàn)樗鼪](méi)有考慮到溶劑化結(jié)構(gòu)。因此,進(jìn)行了分子動(dòng)力學(xué)(MD)模擬以反映電解質(zhì)中的情況。如圖3g和3i所示,電解質(zhì)中的Na+通過(guò)Na···Odiglyme與兩個(gè)DME分子相互作用,而K+通過(guò)K···Odiglyme與三個(gè)DME分子配位。徑向分布函數(shù)(RDFs)表明,Na比K更接近Odiglyme(圖3j),但Na的配位數(shù)(CN)比K的小。這可能是由于K+尺寸比較大。
然后,取溶劑化結(jié)構(gòu)來(lái)計(jì)算最高占有分子軌道(HOMO)和最低未占有分子軌道(LUMO)的能級(jí)。通常情況下,LUMO的能量越低,表明該物種的優(yōu)先還原。[K(diglyme)3]+的LUMO位于-1.79 eV(圖3k),高于[Na(diglyme)2]+(-2.14 eV)。
所以,[Na(diglyme)2]+將率先被還原。這一結(jié)果再次證實(shí)了K+在電解質(zhì)中相對(duì)較好的電化學(xué)穩(wěn)定性。除了熱力學(xué),動(dòng)力學(xué)也阻礙了K+在反應(yīng)中的參與。因?yàn)槿軇┗Y(jié)構(gòu)在還原前需要解離,K+和Na+的溶劑化能可以被認(rèn)為是評(píng)估能量屏障的一個(gè)指標(biāo)。如圖3l所示,[K(diglyme)3]+顯示出比[Na(diglyme)2]+(-4.69 eV)更大的溶劑化能(-24.97 eV)。這些數(shù)據(jù)意味著[K(diglyme)3]+的解離需要比[Na(diglyme)2]+的解離更多能量。
換句話說(shuō),[K(diglyme)3]+比[Na(diglyme)2]+更穩(wěn)定。這一結(jié)論也得到了靜電勢(shì)(ESP)圖的支持。Na(diglyme)2]+的平均ESP比[K(diglyme)3]+的更正,這使得它對(duì)電子攻擊高度活躍,有利于電化學(xué)還原。簡(jiǎn)而言之,高配位數(shù)、大的溶劑化能、高的LUMO能級(jí)和離域電荷分布使得K+在電解質(zhì)中更加穩(wěn)定,這與馬庫(kù)斯理論的預(yù)測(cè)非常一致。
基于這一理論,具有較少溶劑分子和局部電荷分布的巨型結(jié)構(gòu)具有小的重組能壘。在這種情況下,溶劑化的Na+離子比K+離子的對(duì)應(yīng)物更活躍。所有這些結(jié)果在理論上和實(shí)驗(yàn)上都證明,K+不參與Sn的電化學(xué)反應(yīng)。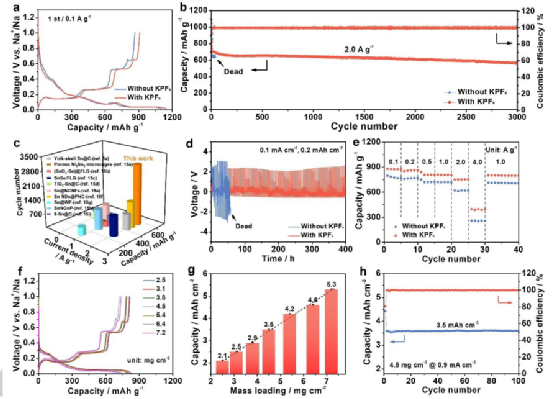
圖4、KPF6增強(qiáng)的電化學(xué)性能。
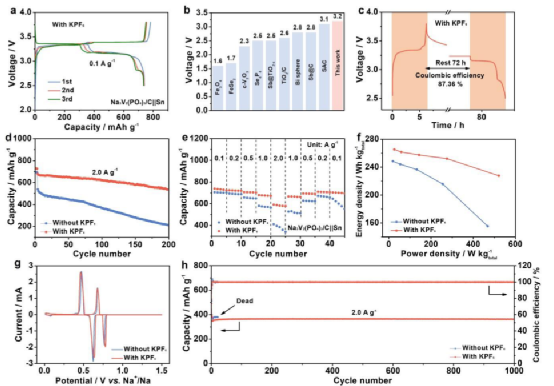
圖5、NVP@C||Sn和Bi||Na的電化學(xué)性能。
總之,K+離子被引入電解質(zhì)以促進(jìn)均勻的反應(yīng)和電極的穩(wěn)定性,這在化學(xué)機(jī)械模型、SEM和AFM中得到了證明。CV曲線、EDS和XPS光譜表明,K+離子不直接參與電化學(xué)反應(yīng),這一點(diǎn)得到了由Nernst方程估計(jì)的氧化還原電位、LUMO的能級(jí)和基于MD模擬得出的溶劑化成分的解離能的支持。 靜電屏蔽的作用使μ-Sn在2 A g-1的條件下經(jīng)過(guò)3000次循環(huán)后能提供565 mAh g-1的容量。
相比之下,在電解液中沒(méi)有K+的情況下循環(huán)的電極只能存活34次。K+帶來(lái)的益處在高負(fù)荷電極、全電池和其他陽(yáng)極材料中也得到了證實(shí),反映了未來(lái)的巨大潛力。這種策略特別耐人尋味,因?yàn)椴恍枰M(fèi)力的準(zhǔn)備、復(fù)雜的儀器和昂貴的試劑。
審核編輯:劉清








 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論