 電子發(fā)燒友App
電子發(fā)燒友App


自誕生以來(lái),GPU就被廣泛應(yīng)用于高性能科學(xué)計(jì)算領(lǐng)域。GPU的大規(guī)模并行計(jì)算能力幫助包括分子模擬在內(nèi)的許多傳統(tǒng)科學(xué)計(jì)算應(yīng)用實(shí)現(xiàn)了爆炸式提速。分子模擬是一種利用計(jì)算機(jī)在微觀尺度下研究材料、蛋白質(zhì)、藥物等分子行為的技術(shù)。由于其模擬流程中包含許多高計(jì)算量的模塊,分子模擬程序非常適合通過(guò)GPU來(lái)加速。經(jīng)過(guò)十幾年的發(fā)展迭代,目前大部分常用的分子模擬軟件都對(duì)GPU硬件進(jìn)行了不同程度的適配。本文將對(duì)分子模擬中關(guān)鍵的近程力計(jì)算模塊在三款不同模擬軟件中的GPU移植案例展開(kāi)深度剖析,希望能以小窺大,探究科學(xué)計(jì)算應(yīng)用在GPU上優(yōu)化的關(guān)鍵點(diǎn)。
在本系列的上篇《分子模擬:打開(kāi)微觀世界的鑰匙(上)—— 起源、發(fā)展與現(xiàn)狀》,我們介紹了分子模擬技術(shù)的起源、發(fā)展與現(xiàn)狀。分子模擬技術(shù)可以通過(guò)一個(gè)體系的化學(xué)組成和其所處的環(huán)境來(lái)預(yù)測(cè)其物理/化學(xué)以及動(dòng)力學(xué)性質(zhì),是科學(xué)計(jì)算領(lǐng)域的一個(gè)重要組成部分。而作為一項(xiàng)歷史悠久的仿真技術(shù),傳統(tǒng)的分子模擬程序通常都是通過(guò)多線程的方法在CPU上進(jìn)行模擬。但在GPU橫空出世以后,人們驚喜地發(fā)現(xiàn),在GPU的超高并行效率的協(xié)助下,分子模擬的速度和模擬體系的尺寸都可以得到數(shù)量級(jí)的提升。因此,目前的大部分主流分子模擬應(yīng)用中都加入了GPU版本供用戶使用。然而,仔細(xì)研究后可以發(fā)現(xiàn),各大應(yīng)用在GPU上的適配方法差異很大。本文將以分子動(dòng)力學(xué)計(jì)算中的近程力計(jì)算模塊(short-range force)為例,拆解幾大主流應(yīng)用中的GPU算法細(xì)節(jié)。本文中提到的GPU術(shù)語(yǔ)皆以CUDA體系為標(biāo)準(zhǔn),包括:thread(線程), warp(線程束), shared memory(共享內(nèi)存), host(主設(shè)備), device(計(jì)算設(shè)備)等。
1
分子動(dòng)力學(xué)近程作用力計(jì)算模塊
本系列上篇曾經(jīng)介紹過(guò),分子動(dòng)力學(xué)模擬中體系力場(chǎng)的計(jì)算可以被大致分為兩大部分:成鍵作用力(bonded interactions)和非成鍵作用力(nonbonded interactions)。顧名思義,成鍵作用力計(jì)算的是互相形成化學(xué)鍵的原子之間的作用力,而非成鍵作用力則是體系中所有沒(méi)有形成化學(xué)鍵的原子間的相互作用力。由于非成鍵作用力的計(jì)算需要遍歷體系中所有可能的原子對(duì),其復(fù)雜度達(dá)到了 O(N2) (此處N為體系中的總原子數(shù))。因此,非成鍵作用力的計(jì)算是分子動(dòng)力學(xué)模擬中最耗時(shí)的部分。考慮到該作用力隨距離的增加而快速衰減的特性,我們可以通過(guò)引入截?cái)嗑嚯x(cutoff distance)的概念來(lái)簡(jiǎn)化計(jì)算。我們將小于截?cái)嗑嚯x的部分作用力稱為近程作用力,而大于截?cái)嗑嚯x的部分則稱遠(yuǎn)程作用力。
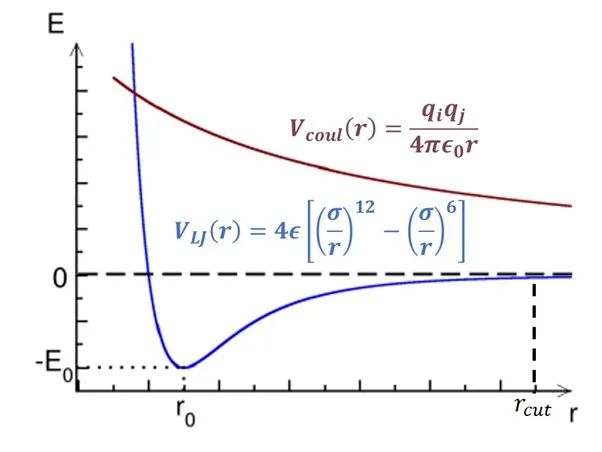
圖1:非成鍵作用勢(shì)隨距離變化簡(jiǎn)圖。
橫軸為距離,縱軸為勢(shì)能。
如圖1所示,范德華勢(shì)(VLJ, 色散作用)和庫(kù)倫勢(shì)(Vcoul, 電荷作用)都隨距離r的增加而遞減。其中范德華勢(shì)在截?cái)嗑嚯x(rcut)處基本衰減到零,因此大部分情況下這部分計(jì)算只需要考慮截?cái)嗑嚯x內(nèi)的原子對(duì)。但庫(kù)倫勢(shì)衰減速度較慢,我們無(wú)法直接忽略其長(zhǎng)程(>rcut)部分。庫(kù)倫勢(shì)的長(zhǎng)程部分可以通過(guò)目前最先進(jìn)的PME(particle-mesh Ewald)方法[1]計(jì)算,該算法的時(shí)間復(fù)雜度僅為O(NlogN)。由此可見(jiàn),近程作用力的計(jì)算(O(N2))是模擬體系力場(chǎng)算法中最消耗計(jì)算資源的部分。如圖2所示,非成鍵作用力中的近程力計(jì)算開(kāi)銷(SR+NEIGH)在總開(kāi)銷中的占比高達(dá)~80%。因此,近程力計(jì)算模塊的加速是提升模擬速度的關(guān)鍵所在,該模塊也成為了最早被移植到GPU上的計(jì)算單元,目前各大主流模擬軟件中都有較為成熟的GPU近程力計(jì)算版本。后面幾個(gè)小節(jié)中,筆者將討論近程力計(jì)算模塊在CPU上和不同軟件的GPU版本中的算法差異。
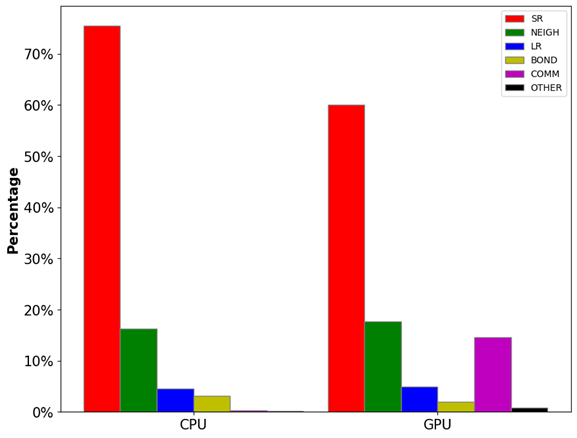
圖2:LAMMPS分子動(dòng)力學(xué)模擬中各模塊的計(jì)算開(kāi)銷占比。其中,SR為近程力計(jì)算開(kāi)銷,NEIGH為鄰近列表構(gòu)建開(kāi)銷,LR為長(zhǎng)程力計(jì)算開(kāi)銷,BOND為成鍵作用力計(jì)算開(kāi)銷,COMM為通訊開(kāi)銷,OTHER為其他開(kāi)銷。
測(cè)試體系:視紫紅質(zhì)(Rhodopsin)蛋白質(zhì)體系,96,000原子,LAMMPS Benchmarks
2
近程力CPU算法
對(duì)于一個(gè)包含N個(gè)原子的體系,計(jì)算近程作用力最直接的方法就是雙循環(huán)遍歷所有可能的原子對(duì),并通過(guò)距離和力場(chǎng)參數(shù)計(jì)算部分作用力,最后對(duì)所有結(jié)果進(jìn)行歸約操作。很明顯,即使在引入截?cái)嗑嚯x以后,此種暴力求解方法的時(shí)間復(fù)雜度仍為O(N2)。因此,我們需要一些技巧來(lái)加速近程力計(jì)算模塊,而neighbor list(鄰近列表)方法就是一種最常用的技巧。neighbor list方法為體系內(nèi)的每一個(gè)原子都構(gòu)建一張列表,其中存儲(chǔ)著在三維空間中與其臨近(距離小于截?cái)嗑嚯x)的其他原子的編號(hào)。當(dāng)這些列表構(gòu)建完畢以后,在計(jì)算近程力時(shí),我們只需要遍歷每個(gè)原子與其neighbor list中的其他原子的作用力即可,因?yàn)榻財(cái)嗑嚯x外的作用力可以忽略不計(jì)。如此一來(lái),近程力計(jì)算的復(fù)雜度就被減小到了O(N)。
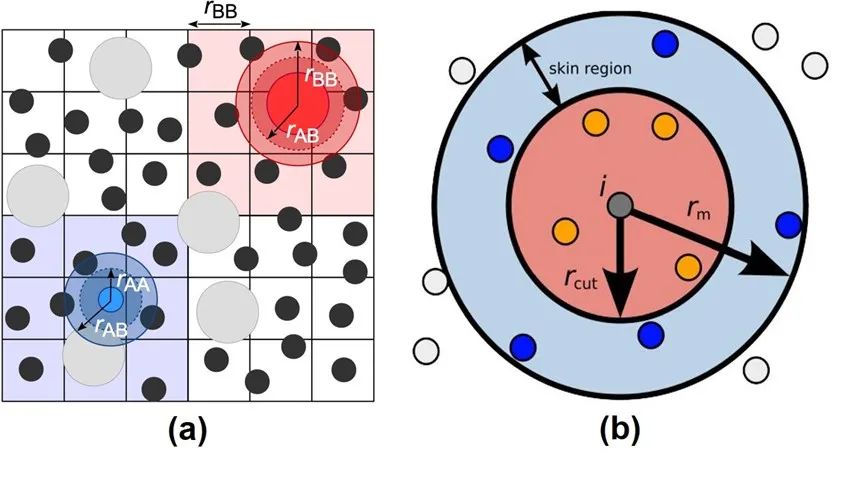
圖3:Verlet Neighbor List方法示意圖。
來(lái)源:Neighbor lists — HOOMD-blue 2.5.2 documentation
然而,構(gòu)建neighbor list的時(shí)間是無(wú)法被忽略的。同時(shí),由于在模擬過(guò)程中原子位置會(huì)持續(xù)發(fā)生變化,這些列表需要頻繁地被重建。于是,人們提出了一種可以將neighbor list重復(fù)使用的技巧: Verlet neighbor list方法[2]。考慮到原子運(yùn)動(dòng)的連續(xù)性以及分子動(dòng)力學(xué)模擬中通常步長(zhǎng)很短等特性,原子的坐標(biāo)在若干步模擬過(guò)后通常不會(huì)改變太大。因此,如圖3(a)所示,我們可以把模擬體系劃分成若干網(wǎng)格, 將每一個(gè)原子歸到格子里, 然后只需要搜尋原子當(dāng)前以及周圍總共27個(gè)(假設(shè)3D體系)網(wǎng)格中的其他原子即可。另外,為了記錄原子進(jìn)入或離開(kāi)neighbor list范圍的情況,我們還可以在截?cái)喾秶酝庠O(shè)置一個(gè)緩沖區(qū)域(skin region), 如圖3(b)所示。假如監(jiān)測(cè)到有大量原子離開(kāi)緩沖區(qū),則需立刻重構(gòu)Verlet list;否則,可以按一定的頻率(一般每20到40步模擬)來(lái)更新list。如此便可以做到Verlet list的復(fù)用,節(jié)省大量計(jì)算開(kāi)銷。圖4展示了CPU中近程力計(jì)算的兩大模塊,neighbor list build (a)和粒子間作用力計(jì)算(b)的簡(jiǎn)單計(jì)算流程。


圖4:近程力CPU計(jì)算模塊:
(a) neighbor list 構(gòu)建, (b) 作用力計(jì)算。
3
近程力LAMMPS-GPU算法
本文后面的篇幅中,我將介紹三款分子動(dòng)力學(xué)應(yīng)用中的近程力GPU算法。首先要介紹的是LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)[3]。LAMMPS 是一款由美國(guó)桑迪亞國(guó)家實(shí)驗(yàn)室(Sandia National Lab)開(kāi)發(fā)的開(kāi)源分子動(dòng)力學(xué)模擬程序包。該程序于1995年第一次發(fā)布,距今已有將近30年的歷史。
LAMMPS的GPU包中對(duì)近程力模塊的移植相對(duì)較為直接,并沒(méi)有對(duì)數(shù)據(jù)結(jié)構(gòu)進(jìn)行大幅改變,只是在最新的版本中加入了更多warp-level指令的使用。首先,LAMMPS-GPU的粒子間作用力的計(jì)算基本與CPU版本(見(jiàn)圖4(b))完全一致。簡(jiǎn)單來(lái)說(shuō),即GPU中每一個(gè)thread負(fù)責(zé)一個(gè)原子,循環(huán)計(jì)算該原子與其neighbor list內(nèi)的原子的相互作用力,再將結(jié)果寫(xiě)入到儲(chǔ)存作用力的數(shù)組中。而在neighbor list構(gòu)建模塊中,LAMMPS的GPU版本與CPU版本最大不同是通過(guò)warp shuffle的指令來(lái)減少?gòu)膆ost到device的數(shù)據(jù)讀取。具體來(lái)說(shuō),該算法分為以下步驟:

圖5:LAMMPS-GPU neighbor list build模塊計(jì)算流程。
得益于GPU的高度并行能力以及warp 內(nèi)部的數(shù)據(jù)共享能力,LAMMPS的GPU版本確實(shí)比CPU提升了不少速度。然而,這種直接的GPU移植方式還是存在著許多的缺陷,包括:
01
沒(méi)有改變傳統(tǒng)neighbor list的結(jié)構(gòu),LAMMPS的neighbor list 數(shù)據(jù)結(jié)構(gòu)如圖6所示,每個(gè)粒子都有一個(gè)自己的neighbor list,導(dǎo)致有很多的neighbors被重復(fù)儲(chǔ)存。使用這樣的數(shù)據(jù)結(jié)構(gòu)會(huì)大幅增加host-device間的數(shù)據(jù)傳輸壓力。
02
網(wǎng)格是通過(guò)空間坐標(biāo)均勻劃分的,當(dāng)處理非均質(zhì)體系時(shí)各warp需要處理的粒子數(shù)差異較大,易造成線程分歧(thread divergence)。
03
計(jì)算粒子間作用力時(shí)沒(méi)有利用好shared memory/warp shuffle功能來(lái)減少?gòu)膆ost到device的數(shù)據(jù)傳輸。
由此,我們可以看到,想要更好地激發(fā)GPU的潛力,程序還需要對(duì)底層的數(shù)據(jù)結(jié)構(gòu)和運(yùn)算邏輯進(jìn)行進(jìn)一步優(yōu)化。
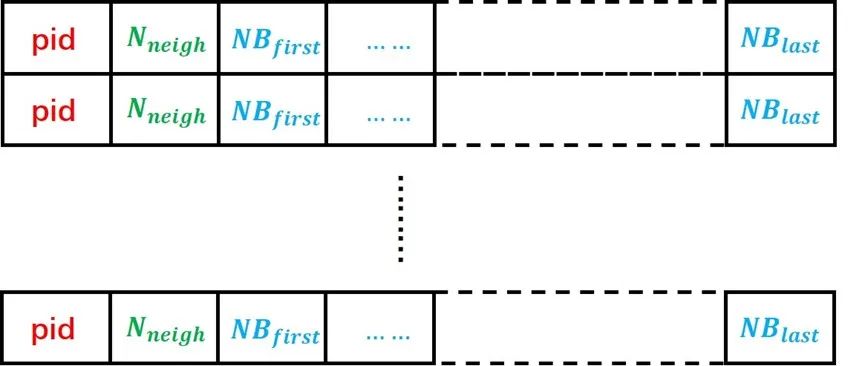
圖6:LAMMPS中的neighbor list數(shù)據(jù)結(jié)構(gòu)。每個(gè)原子對(duì)應(yīng)其中的一行數(shù)據(jù),pid為原子編號(hào),Nneigh為該原子的neighbor總數(shù),NBfirst至NBlast為從第一個(gè)neighbor到最后一個(gè)neighbor的編號(hào)。
4
近程力GROMACS-GPU算法
GROMACS(GROningen MAchine for Chemical Simulations)[4]是一款由荷蘭格羅寧根大學(xué)團(tuán)隊(duì)開(kāi)發(fā)的模擬程序,該軟件于1991年首次發(fā)布,目前已經(jīng)成為全球使用人數(shù)最多的分子動(dòng)力學(xué)軟件之一。GROMACS從2013年的4.5版本開(kāi)始支持GPU,經(jīng)過(guò)多次迭代優(yōu)化之后,目前的最新版本已經(jīng)可以在GPU上完成全模擬流程。
與LAMMPS-GPU不同的是,GROMACS的開(kāi)發(fā)團(tuán)隊(duì)意識(shí)到了傳統(tǒng)的neighbor list結(jié)構(gòu)不適合GPU的SIMT(單指令多線程)計(jì)算特性。因此,他們提出了一種基于cluster(聚類)結(jié)構(gòu)的新型近程力算法[5]。受到矩陣乘法運(yùn)算的啟發(fā),該方法通過(guò)將體系中的粒子分為N x M大小的cluster來(lái)提升GPU的計(jì)算效率。
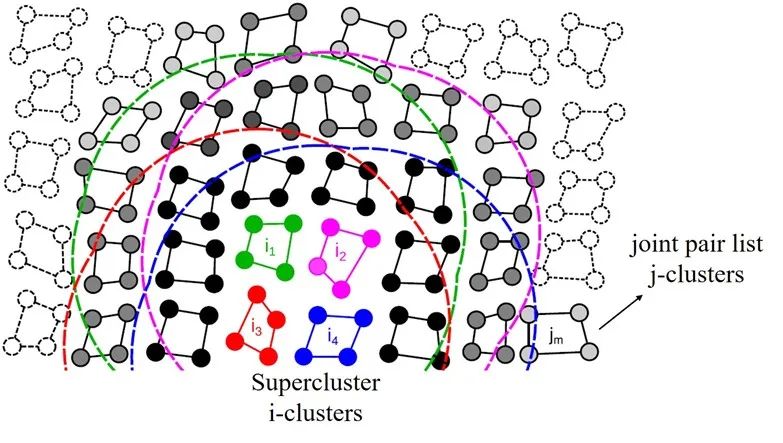
圖7:GROMACS-GPU算法中的super-cluster結(jié)構(gòu)。圖中的super-cluster包含i1-i4 四個(gè)i-cluster,這些i-cluster的neighbors被整合成了一個(gè)joint pair list, 即圖中黑色和灰色的j-cluster合集。
來(lái)源:Páll etc. “Heterogeneous parallelization and accelerationof molecular dynamics simulations in GROMACS”, J. Chem. Phys. 153, 134110 (2020)
以圖7為例,圖中的體系被分為包含N=4個(gè)粒子的i-cluster和包含M=4個(gè)粒子的j-cluster。不同于以單個(gè)粒子為計(jì)算單元的傳統(tǒng)neighbor list算法,此新方法以粒子的聚類為單位來(lái)構(gòu)建neighbor list。具體來(lái)說(shuō),在搜索neighbor的過(guò)程中,程序會(huì)遍歷所有的i-cluster,并找到在其截?cái)嗑嚯x范圍內(nèi)的j-cluster加入其neighbor list。如此一來(lái),在計(jì)算作用力的時(shí)候就可以每次同時(shí)載入M?個(gè)粒子的信息,從而減少對(duì)neighbor數(shù)據(jù)的重復(fù)讀取操作。盡管這種做法會(huì)無(wú)可避免的增加一些無(wú)效計(jì)算(并不是cluster中的每個(gè)粒子都在目標(biāo)粒子的截?cái)喾秶鷥?nèi)),但是對(duì)于這樣一個(gè)瓶頸在內(nèi)存操作的程序來(lái)說(shuō),使用該方法的收益還是遠(yuǎn)遠(yuǎn)大于損失。
在實(shí)際應(yīng)用中,這種聚類臨近列表(cluster neighbor list)方法可以進(jìn)行進(jìn)一步的優(yōu)化來(lái)占滿目標(biāo)硬件的執(zhí)行寬度(execution width)。以Nvidia的GPU產(chǎn)品為例,N卡以warp為計(jì)算單元,每個(gè)warp中包含32個(gè)thread,因此其執(zhí)行寬度為32。我們可以通過(guò)將cluster的大小設(shè)置為N=8,M=4 來(lái)達(dá)到最高計(jì)算效率。同時(shí),還可以將4個(gè)i-cluster合并成一個(gè)super-cluster(見(jiàn)圖7中的i1-i4), 并將它們的neighbor list整合成一個(gè)聯(lián)合成對(duì)列表(joint pair list)。如此操作的好處在于可以充分利用GPU中較為快速的shared memory來(lái)進(jìn)一步減少host到device端的數(shù)據(jù)傳輸。在每一次計(jì)算作用力時(shí),程序會(huì)先將super-cluster中所有粒子的數(shù)據(jù)讀取進(jìn)來(lái)并儲(chǔ)存在shared memory中,這樣就避免了在后續(xù)的計(jì)算過(guò)程中進(jìn)行緩慢的顯存讀取(global memory load)操作。同理,在執(zhí)行寬度為64的AMD GPU上,需要設(shè)置N=M=8 ; 而在英特爾硬件上則需將cluster的大小設(shè)置為N=4,M=2 。
通過(guò)對(duì)底層數(shù)據(jù)結(jié)構(gòu)的改進(jìn)優(yōu)化,GROMACS成功提升了GPU計(jì)算資源的利用效率。與傳統(tǒng)的neighbor list結(jié)構(gòu)相比,cluster pair list的使用可以顯著減少同一個(gè)neighbor的重復(fù)儲(chǔ)存,在數(shù)據(jù)復(fù)用和內(nèi)存使用等方面做到了很好的優(yōu)化。同時(shí),相對(duì)均勻的計(jì)算分配也可以大幅縮短thread的閑置和等待時(shí)間。盡管增加了一定的計(jì)算量,但是在這個(gè)內(nèi)存瓶頸的程序中,這部分增加的計(jì)算時(shí)間可以被隱藏在內(nèi)存操作延遲中,從而不對(duì)整體性能產(chǎn)生很大影響。因此,筆者認(rèn)為GROMACS-GPU的近程力計(jì)算模塊是一個(gè)很成功的GPU移植和優(yōu)化案例。
5
近程力OpenMM-GPU算法
接下來(lái)要介紹的OpenMM[6]是一款主打易用性、拓展性和高性能的新型模擬引擎。該軟件的開(kāi)發(fā)團(tuán)隊(duì)主要來(lái)自斯坦福大學(xué),他們?cè)O(shè)計(jì)這款軟件的初衷就是想要解決傳統(tǒng)分子模擬軟件難上手、源碼修改困難以及在GPU上性能不佳等問(wèn)題。經(jīng)過(guò)近十年的努力,OpenMM也確實(shí)越來(lái)越受學(xué)者們的歡迎。一方面,OpenMM的Python 和C++ API都做的非常完善,用戶可以自由添加新功能或者修改現(xiàn)有功能。另一方面,OpenMM的GPU算法先進(jìn),加速效果非常好。因此,許多新的研究項(xiàng)目都選擇其作為模擬引擎,包括著名的蛋白質(zhì)結(jié)構(gòu)預(yù)測(cè)模型Alpha Fold[7]。
為了提升近程力計(jì)算在GPU上的性能,OpenMM同樣在neighbor list的數(shù)據(jù)結(jié)構(gòu)上進(jìn)行了優(yōu)化。其整體的優(yōu)化思路與GROMACS比較類似,也是通過(guò)將多個(gè)粒子集合成atom block(原子塊)的方式來(lái)構(gòu)建neighbor list,以減少neighbors的重復(fù)儲(chǔ)存和數(shù)據(jù)傳輸。兩種方法的最大區(qū)別在于每個(gè)集群中粒子數(shù)量的選擇。OpenMM的atom block中包含32或64個(gè)原子,而GROMACS中的cluster則只有4或8個(gè)粒子。增加集群的尺寸可以減少neighbor list搜索和構(gòu)建的開(kāi)銷,因?yàn)檫@部分計(jì)算的耗時(shí)是由體系中的集群總數(shù)決定的。但同時(shí),大集群也意味著在作用力計(jì)算模塊中會(huì)引入更多的無(wú)效計(jì)算,增加計(jì)算資源的浪費(fèi)。因此,集群尺寸的選取實(shí)際上是近程力計(jì)算中兩大模塊之間的平衡取舍問(wèn)題。OpenMM團(tuán)隊(duì)經(jīng)過(guò)驗(yàn)證后發(fā)現(xiàn)較大的集群在現(xiàn)代GPU架構(gòu)中可以更為高效。同時(shí),他們還使用了一些技巧來(lái)降低增大集群尺寸后引入的負(fù)面效應(yīng)。比如,在構(gòu)建neighbor list的時(shí)候增加一個(gè)細(xì)致搜索的步驟。
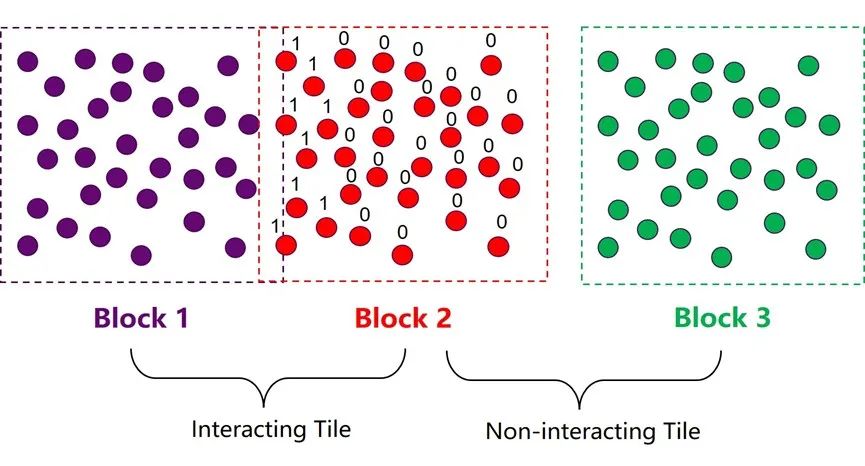
圖8:OpenMM-GPU算法中的atom block 和tile結(jié)構(gòu)。
如圖8所示,在第一步粗略搜索的過(guò)程中,程序會(huì)將邊界有交集的兩個(gè)atom block 標(biāo)記為相互作用集群對(duì)(interacting tile);邊界沒(méi)有交集的atom block對(duì)則被標(biāo)記為互不作用集群對(duì) (non-interacting tile)。這一步搜索結(jié)束以后,程序會(huì)馬上進(jìn)入下一步細(xì)致搜索階段,這一步的目的是構(gòu)建一個(gè)32/64位的mask,對(duì)block 2中的所有粒子進(jìn)行判定,假如一個(gè)粒子與block 1中的任意粒子有相互作用,則將該位設(shè)為1,反之則設(shè)為0。這個(gè)mask中儲(chǔ)存的信息可以有效規(guī)避后續(xù)作用力模塊中的大部分冗余計(jì)算。OpenMM中這種類似的小trick還有很多,正是因?yàn)樗麄儗?duì)GPU更深的理解和對(duì)細(xì)節(jié)的注重,使得OpenMM在GPU上的加速效果達(dá)到了業(yè)界最頂尖的水平。
結(jié)語(yǔ)
本文介紹了三款常用的分子動(dòng)力學(xué)模擬應(yīng)用中的GPU加速算法,分別代表三種不同程度的GPU適配思路。其中,LAMMPS的GPU版本基本直接沿用了CPU上的算法,只是利用GPU的高度并行能力對(duì)其中的計(jì)算密集部分進(jìn)行加速。而GROMACS和OpenMM則將CPU版本中的底層數(shù)據(jù)結(jié)構(gòu)和計(jì)算邏輯進(jìn)行了大刀闊斧地改造,以更好地發(fā)揮GPU的硬件特性。另外,OpenMM通過(guò)一些算法細(xì)節(jié)上的巧妙設(shè)計(jì)進(jìn)一步提升了程序在GPU上的運(yùn)行效率。
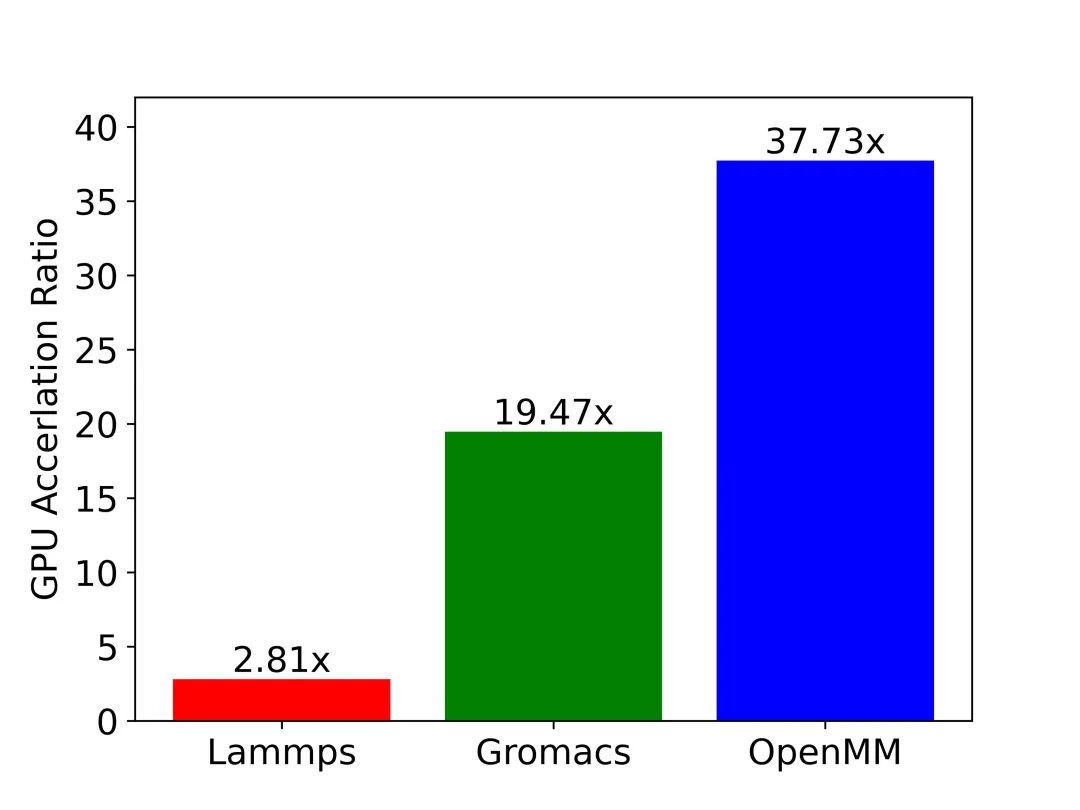
圖9:三大應(yīng)用GPU加速效果一覽。圖中縱軸為GPU相對(duì)CPU的加速比。測(cè)試用例:包含45,000原子的SPC/E模型水體系,通過(guò)沐曦MetaMol分子模擬工作流軟件實(shí)現(xiàn)相同體系在不同應(yīng)用上的模擬。
測(cè)試硬件:CPU: AMD Ryzen 7 3700X 8-Core Processor; ?GPU: NVIDIA GeForce RTX 3080。
圖9中展示了LAMMPS、GROMACS和OpenMM在模擬SPC/E模型水體系時(shí)的GPU-CPU的加速比數(shù)據(jù)。測(cè)試結(jié)果與前文中的分析一致:在相同的硬件上模擬同一體系時(shí),OpenMM的GPU加速效率要遠(yuǎn)高于GROMACS和LAMMPS。讀到這里大家應(yīng)該都有疑問(wèn):為什么LAMMPS不對(duì)數(shù)據(jù)結(jié)構(gòu)進(jìn)行優(yōu)化呢?究其原因,以LAMMPS為代表的傳統(tǒng)軟件歷史悠久,在設(shè)計(jì)之初并沒(méi)有考慮到在不同平臺(tái)、不同硬件上適配的需求,導(dǎo)致軟件的移植工作量巨大,而修改底層數(shù)據(jù)結(jié)構(gòu)更是牽一發(fā)而動(dòng)全身,幾乎難以完成。另一方面,像OpenMM這種近年來(lái)開(kāi)發(fā)的新型應(yīng)用則對(duì)軟件的拓展性和靈活性非常重視。比如在OpenMM中,用戶可以很便捷地通過(guò)插件的形式來(lái)添加新的計(jì)算平臺(tái)或是新算法。因此,在各種硬件架構(gòu)層出不窮,日新月異的今天,如何保證拓展性和靈活性需要成為我們開(kāi)發(fā)科學(xué)計(jì)算軟件時(shí)必須考慮的問(wèn)題。
個(gè)人認(rèn)為這些軟件的GPU移植和優(yōu)化的實(shí)例比較具有研究?jī)r(jià)值,希望這篇文章可以給各位讀者帶來(lái)一些啟發(fā),拓寬一些思路。本文中的大部分內(nèi)容都來(lái)自筆者自己對(duì)軟件源碼的研究和模擬數(shù)據(jù)的分析所得。文中提出的觀點(diǎn)都來(lái)自我的個(gè)人理解,難免有所紕漏,歡迎大家批評(píng)指正。最后,希望通過(guò)進(jìn)一步的優(yōu)化和改進(jìn),我們可以讓分子模擬在沐曦的通用GPU上達(dá)到更高的速度和精度,真正成為人類打開(kāi)微觀世界的鑰匙。
編輯:黃飛













 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論